Nav1.7
Description: sodium channel, voltage-gated, type IX, alpha Gene: Scn9a Alias: nav1.7, scn9a, PN1, ETHA, NENA, FEB3B, NE-NA, GEFSP7
Nav1.7, encoded by the gene scn9a, is a sodium, voltage-gated, type 9, alpha subunit channel. Nav1.7 is mainly expressed in the PNS. The channels plays a significant role in nociception signaling by regulating sensory neuron excitability. have been associated with the deregulation of channel excitability and lead to a number of pain disorders
Experimental data
Mouse Nav1.7 gene in CHO host cells |
||
|
Click for details 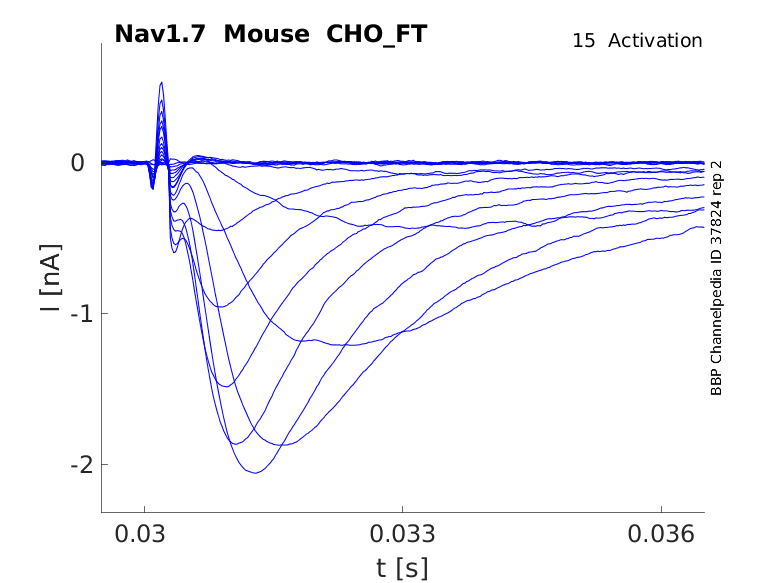
15 °Cshow 40 cells |
Click for details 
25 °Cshow 48 cells |
Click for details 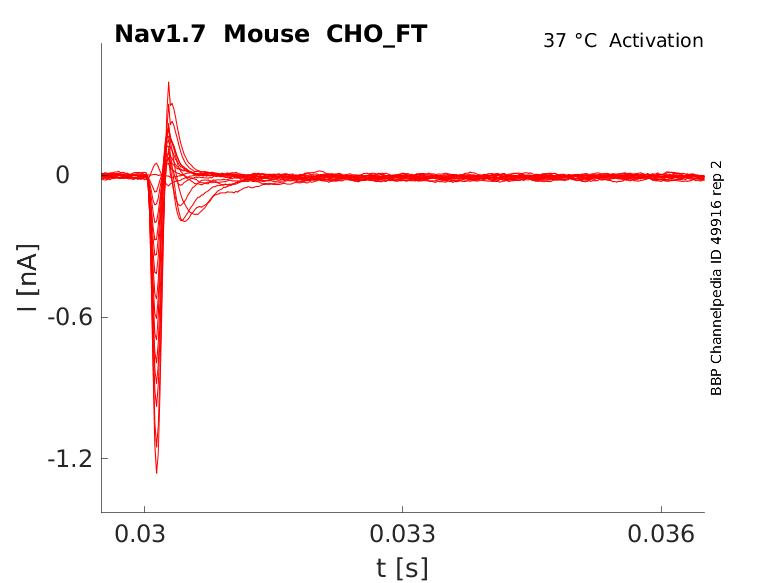
35 °Cshow 52 cells |
In humans, scn9a, the gene which encodes Nav1.7, is composed of 27 exons from which 26 are coding (exon1 is non-coding) spanning 167.3 Mb on chromosome 2 (2q24). [2202]
There exist multiple Nav1.7 transcript variants across species as a result of the alternative splicing of scn9a (see Protein Isoform table).
Notably, like other Navs, the alternative splicing of exon 5 results in a “neonatal” variant of Nav1.7. However, though the gene splicing event is similar to that of other neonatal variants occurring in VGSC, Nav1.7 exon 5N is not developmentally regulated. Instead, scn9a exon 5N is preferentially expressed in the PNS and CNS of adult tissues whereas significant usage of exon 5A was found only in DRG.
Additionally, there exists alternative 5′-splice donor sites in exon 12 (coding exon 11), which encode for an extended segment of the cytoplasmic loop between domains I and II. [2202] [2203]
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_002977.3 | 9771 | |
| Mouse | NM_001290674.1 | 9865 | |
| Rat | NM_133289.2 | 9859 |
The human Nav1.7 protein is composed of 1988 amino acid (aa) and has a molecular weight of 226 Kda.
There exists a number of protein isoforms that arise from the translation of the aforementioned transcript variants:
- Isoforms that are translated from transcript exon 5 N differ by 2 residues only from the canonical sequence.
- Isoforms resulting from the translation of transcript with exon 12 lead to an elongation of exon 11, which extends the extracellular loop 1 (L1) between domain 1 and 2 (DI-DII). The two forms of L1 are designated L1‐short (S) and L1‐long (L).
These isoforms not only vary in final protein length and structure but also in expression pattern. For example, Isoform 1 is expressed preferentially in the central and peripheral nervous system whereas isoform 2 is preferentially expressed in the dorsal root ganglion [2202].
Some isoform expression patterns are altered as a result of disease. It was shown that Nav1.7 11S was upregulated and contributed to pain hypersensitivity in animal models of neuropathic pain [2128]
Isoforms
Nav1.7 hosts a number of glycosylation sites, indicating that the protein is likely glycosylated [2205]. However, the exact timing of glycosylation and its impacts on channel kinetics or expression have not been extensively studied. Research has shown that Nav1.7 can exist in at least two different glycosylated states in HEK293 cells: a heavily functional glycosylated form and a core-glycosylated immature form [2128].
Nav1.7 is, however, subject to phosphorylation by various enzymes [2128]
- Protein Kinase A (PKA) has been shown to reduce peak current in Nav1.7 cell expression systems. PKA’s presence has been shown to increase after inflammation and in pathological pain along with a reduction in Nav1.7 expression. However, it demonstrated that PKA phosphorylation of the Nav1.7 11S isoform exhibited activation curves that were shifted to hyperpolarized potentials, thus presumably increasing neuronal excitability. Other studies explain that these differences in PKA’s impact on Nav regulation vary depending on the cell type. Therefore, though PKA phosphorylation does affect Nav1.7, careful characterization of its effects are required to determine its activity in specific locations
- During an inflammatory response, PKB activation is shown to upregulate Nav1.7 expression
- PKC phosphorylation acts similarly to PKA as it leads to an increase in Nav1.7 sodium current in certain cell types and decrease in channel expression in others.
- Certain members of the MAPK family phosphorylate Nav1.7, making it open at lower thresholds of activation. This type of phosphorylation is often present within the context of acquired pain conditions
Nav1.7 is subject to ubiquitination. The channel contains a PY motif that was shown to be negatively regulated by Nedd4-2. Knockout experiments confirmed the role of Need4-2 in Nav1.7 ubiquitination as downregulation of the ubiquitin ligase led to an increase in Nav1.7 expression. [2128]
Visual Representation of Nav1.7 Structure
Methodology for visual representation of structure available here

Like all voltage gated sodium channels, Nav1.7 is made up of a single protein comprised of 4 homologous domains (DI-DIV). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.7 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus deactivating the channel. Nav1.7 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of human Nav1.7 was resolved via cryo-electron microscopy, at a 2.2-Å resolution, giving us a detailed insight into the channel’s architecture. The structure of Nav1.7 is almost identical to that of Nav1.4, namely having selectivity filter asymmetry and an up conformation for voltage sensing domains S1-S4. Another characteristic of Nav1.7’s structure is that each S6 segment is followed by a cytosolic helix. Each of these 4 helices have a distinct conformation. S6-D1 is exceptionally long and extends directly out into the cytosol. S6-DII is followed by the transverse II-III helix. S6-DIII is succeeded by the short III-IV helix, and S6-DIV is connected to the globular C-terminal domain (CTP). These cytosolic segments further contribute to the structural asymmetry of the pore domain [2206]
The approximative size/surface of Nav1.5 can be determined via the resolved or predicted structures.
Nav1.7 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Nav1.7 is a fast activating and inactivating channel that conducts a large inward Na+ current following depolarization. However, compared to other voltage gated sodium channels, Nav1.7 recovers slower from fast inactivation. Nav1.7 is also characterized by a slow closed-state inactivation, allowing it to produce a ramp current in response to small depolarizations [2207]
Single channel unitary conductance
Single channel unitary conductance is determined experimentally. For Nav1.7, there have been recordings of single channel unitary conductance values with some degree of variance between experiments:
- 19.5pS [2271]
Model
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Species : Human | Gene: scn9a (+ β1)
Host cell: tsa201 (HEK293) | Temperature: RT (to 25 C by Q10)
Formalism: Markov | States: C1, C2, O1, O2, I1, I2
Implementation: NEURON | Simulation: Nav17_a.mod

Kinetic Modeling of Nav1.7 Provides Insight Into Erythromelalgia-associated F1449V Mutation (Gurkiewicz et al, 2011)
https://modeldb.science/138082?tab=1
Species: Human | Gene: Scn9a
Host cell: HEK293 | Temperature: RT
Formalism: Markov | States: C1, C2, C3, O, I1, I2
Implementation: NEURON | Simulation: Nav17WTNaG3e.mod

Membrane Systems Group 2023
Species: Mouse | Gene: Scn9a
Host cell: CHO | Temperature: 25 C
Formalism: Hodgkin-Huxley | Gates: m3, h
Implementation: NEURON | Simulation: Nav17__mCHO25c.mod

Cellular and Tissue
Nav1.7 is preferentially expressed in the peripheral nervous system (PNS) in areas such as [1419](9037087)(21118538):
- Somatosensory ganglion neurons
- Sympathetic ganglion neurons
- Myenteric neurons
- Olfactory sensory neurons (OSN)
- Nodose ganglion neurons
- Visceral sensory neurons
- Smooth myocytes
- Small & large diameter DRG neurons (both Aβ-fibres and C-fibres)
Nav1.7 is known to be expressed in the central nervous system (CNS) in human and monkey spinal cord and brain, although at much lower level [2204]
Developmental
Nav1.7 is predominantly expressed in the postnatal and adult PNS
The subcellular distribution of Nav1.7 depends on the cell type in which it is present.
NaV1.7 is uniformly distributed in the somata in both large and small diameter DRG neurons and along the identified Aβ-fibers and C fibers, being expressed at high levels in nociceptive neurons [850]. In the DRG and trigeminal ganglion neurons, it can be found at the neurite tips [2207]
Given its ability to boost subthreshold stimuli and thus increase the probability of action potential firing, Nav1.7 is often considered as a threshold channel. However, Nav1.7's contribution to other physiological processes is increasingly being recognized.
Sensory Modalities
As Nav1.7 is expressed in a number of sensory neurons, the channel is involved in multiple sensing modalities [2202]:
- Olfaction: Nav1.7 is the predominant sodium channel present in the presynaptic OSNs. Knock-out scn9a mice were shown to be anosmic and unable to suckle, leading to subsequent death. This evidence highlights the role of Nav1.7 in olfaction.
- Cough reflex: itchy “urge-to-cough” is mediated by the stimulation of nodose ganglion neurons, specifically in the C-fiber. Experimental data suggests that Nav1.7 is the main contributor to sodium current in these neurons and Nav1.7 knockdown experiments lead to a reduction of firing in Aβ-fibers and C fibers.
- Acids sensing: Nav1.7 is thought to play a role in acid sensing as modifications, mimicking orthologs that are insensitive to acid induced pain, lead to a loss of action potential firing when exposed to acidic conditions.
Nociception and pain disorders
Nav1.7’s most well-known functionality is its role in pain perception. Indeed, Nav1.7 is present in 85% of all functional nociceptors [2202]. Moreover, blocking of Nav1.7 activity, via the administration of tetrodotoxin (TTX), improved pain symptoms in rats, serving as further proof of the channel’s involvement in pain signalling. [2208]
Given Nav1.7’s important role in the dynamic regulation of nociceptor excitability, deregulation of the channel generally leads to pain related disorders.
Acquired pain conditions
The channel’s contribution to pain has been particularly well studied in the context of animal studies on inflammatory pain. Nav1.7 channel density is dramatically increased in the presence of inflammation [2209] and at least some forms of nerve injury [2210].
Experimental studies note increased amplitudes of TTX-sensitive current following peripheral-tissue inflammation. This increase has been correlated with an increase in both scn9a transcript levels and Nav1.7 protein expression, much more than any other sodium channel involved in pain (nav1.3) [2207]
Inherited pain disorders
There exists a number of pain related channelopathies caused by mutations that alter the activity of Nav1.7. These inheritable mutations generally lead to increased an sensation of pain as a result of channel overexcitability or pain insensitivity, depending on the type of mutation.
- Gain of function: gain of function mutation are dominantly inherited that caused increased pain perception. Three prominent disorders are inherited erythromelalgia (IEM), paroxysmal extreme pain disorder (PEPD), and idiopathic small-fiber neuropathy (I-SFN)
- IEM associated mutations often lead to changes in Nav1.7 by lowering the channel activation threshold and increasing ramp current and slow deactivation. These changes in electrophysiological activity increase neuron excitability [1419].
- PEPD associated mutations lead to channels with incomplete inactivation, enabling a persistent sodium current, as the voltage-dependance of steady-state fast inactivations is shifted towards more depolarizing potentials. [2211] [1419]
- I-SFN associated mutations lead to channel hyperexcitability caused by a number of modifications: depolarized fast inactivation and/or slow inactivation, or an increase in resurgent current [2212] [1419]
- Loss of function: these mutations are recessively inherited and are linked to a complete insensitivity to pain (CIP). Patients with CIP do not experience pain even when subjected to extremely painful stimuli: bone fractures, dental extractions, burns, etc.[1424]
Other de novo mutations have also been clinically identified, like other pain disorders, they cause neuron hyperexcitability and afflicted patients may present mixed/ overlapping IED and PEPD symptoms [2213][1419].
Other disease
Despite only being present at low levels in the CNS, Nav1.7 mutants have been linked to epilepsy/dravet syndromes. However, the mechanisms by which scn9a mutations cause epilepsy are poorly understood [2214].
Deregulation of Nav1.7 has been linked to increased invasiveness of cancerous cells. Indeed blocking of channel functional expression by disrupting agents (TTX, naringenin, si-RNA) lead to a significant loss in cancerous cells mobility [2215] [2216].
Nav1.7 is TTX sensitive
Nav1.7, like other voltage-gated sodium channels, is regulated by various auxiliary proteins and secondary messengers. These regulatory proteins play important roles in the development, localization, and expression of the channel protein. Examples of interacting auxiliary proteins are [2217]:
- Homer2
- Phosphatidylethanolamine-binding protein 1 (PEBP1)
- Fatty acid binding protein 7 (FABP7)
- Mitogen-activated protein (MAP) kinases
- Filamin A
- Microtubule-associated protein 2 (MAP2)
- Calretinin
- Neurotrimin
- Neurofascin
- Fibroblast growth factor 13 (FGF13)
- Multiple PDZ domain protein (MPDZ) and PDZD2
- Collapsin response mediator protein 2 (CRMP2)
- Calmodulin
The binding of Nav β subunits to Nav1.7 regulates the neuronal function of the channel. In the ER/Golgi, Navβ1 and Navβ3 bind to Nav1.7 and help channel glycosylation and proper membrane expression. Navβ2 was shown to be important for the trafficking of the protein.
- Known and predicted drug interactions with Nav1.7
- Known and predicted animal toxin interactions with Nav1.7
References
Herzog RI
et al.
Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons.
J. Physiol. (Lond.),
2003
Sep
15
, 551 (741-50).
Cheng X
et al.
Mutation I136V alters electrophysiological properties of the Na(v)1.7 channel in a family with onset of erythromelalgia in the second decade.
,
2008
, 4 (1).
Brackenbury WJ
et al.
Nerve growth factor enhances voltage-gated Na+ channel activity and Transwell migration in Mat-LyLu rat prostate cancer cell line.
J. Cell. Physiol.,
2007
Mar
, 210 (602-8).
Dib-Hajj SD
et al.
Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable.
,
2008
, 4 (37).
Catterall WA
From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels.
Neuron,
2000
Apr
, 26 (13-25).
Weiss J
et al.
Loss-of-function mutations in sodium channel Nav1.7 cause anosmia.
Nature,
2011
Apr
14
, 472 (186-90).
Gould HJ
et al.
A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain.
Brain Res.,
2000
Jan
31
, 854 (19-29).
Djouhri L
et al.
Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein.
J. Physiol. (Lond.),
2003
Jan
15
, 546 (565-76).
Cummins TR
et al.
Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel.
J. Neurosci.,
1998
Dec
1
, 18 (9607-19).
Toledo-Aral JJ
et al.
Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons.
Proc. Natl. Acad. Sci. U.S.A.,
1997
Feb
18
, 94 (1527-32).
Waxman SG
et al.
Erythermalgia: molecular basis for an inherited pain syndrome.
,
2005
Dec
, 11 (555-62).
Diss JK
et al.
Expression profiles of voltage-gated Na(+) channel alpha-subunit genes in rat and human prostate cancer cell lines.
Prostate,
2001
Aug
1
, 48 (165-78).
Saleh S
et al.
Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes.
J. Physiol. (Lond.),
2005
Oct
1
, 568 (155-69).
Dib-Hajj SD
et al.
The Na(V)1.7 sodium channel: from molecule to man.
Nat. Rev. Neurosci.,
2012
Dec
12
, 14 (49-62).
Dib-Hajj SD
et al.
Sodium channels in normal and pathological pain.
Annu. Rev. Neurosci.,
2010
, 33 (325-47).
Chattopadhyay M
et al.
Continuous delta-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy.
J. Neurosci.,
2008
Jun
25
, 28 (6652-8).
Stamboulian S
et al.
ERK1/2 Mitogen-Activated Protein Kinase Phosphorylates Sodium Channel Nav1.7 and Alters Its Gating Properties.
J. Neurosci.,
2010
Feb
3
, 30 (1637-47).
Ahmad S
et al.
A stop codon mutation in SCN9A causes lack of pain sensation.
Hum. Mol. Genet.,
2007
Sep
1
, 16 (2114-21).
Greenbaum L
et al.
Contribution of genetic variants to pain susceptibility in Parkinson disease.
Eur J Pain,
2012
Oct
, 16 (1243-50).
Shields SD
et al.
Sodium channel Na(v)1.7 is essential for lowering heat pain threshold after burn injury.
J. Neurosci.,
2012
Aug
8
, 32 (10819-32).
Nassar MA
et al.
Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8.
,
2005
, 1 (24).
Nassar MA
et al.
Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain.
Proc. Natl. Acad. Sci. U.S.A.,
2004
Aug
24
, 101 (12706-11).
Laedermann CJ
et al.
Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes.
Front Pharmacol, 2015, 6 (263).
Dib-Hajj SD
et al.
Genetics and molecular pathophysiology of Na(v)1.7-related pain syndromes.
Adv. Genet.,
2008
, 63 (85-110).
Raymond CK
et al.
Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia.
J. Biol. Chem.,
2004
Oct
29
, 279 (46234-41).
Cox JJ
et al.
An SCN9A channelopathy causes congenital inability to experience pain.
Nature,
2006
Dec
14
, 444 (894-8).
Shen H
et al.
Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins.
Science, 2019Mar22, 363 (1303-1308).
Huang G
et al.
High-resolution structures of human Nav1.7 reveal gating modulation through α-π helical transition of S6IV.
Cell Rep, 2022Apr26, 39 (110735).
Dib-Hajj SD
et al.
From genes to pain: Na v 1.7 and human pain disorders.
Trends Neurosci.,
2007
Nov
, 30 (555-63).
Lyu YS
et al.
Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model.
Brain Res, 2000Jul14, 871 (98-103).
Gould HJ
et al.
Rapid sodium channel augmentation in response to inflammation induced by complete Freund's adjuvant.
Brain Res.,
1998
Aug
17
, 802 (69-74).
Liu C
et al.
Nav1.7 protein and mRNA expression in the dorsal root ganglia of rats with chronic neuropathic pain.
Neural Regen Res, 2012Jul15, 7 (1540-4).
Fertleman CR
et al.
SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes.
Neuron,
2006
Dec
7
, 52 (767-74).
Huang J
et al.
Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons.
J. Neurosci.,
2013
Aug
28
, 33 (14087-97).
Estacion M
et al.
NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders.
J. Neurosci.,
2008
Oct
22
, 28 (11079-88).
Singh NA
et al.
A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome.
PLoS Genet.,
2009
Sep
, 5 (e1000649).
Campbell TM
et al.
Functional expression of the voltage-gated sodium channel, Nav1.7, underlies epidermal growth factor-mediated invasion in human [R1.S1] non-small cell lung cancer cells.
J. Cell. Sci.,
2013
Aug
28
, ().
Gumushan Aktas H
et al.
Naringenin inhibits prostate cancer metastasis by blocking voltage-gated sodium channels.
Biomed Pharmacother, 2018Oct, 106 (770-775).
Chew LA
et al.
Mining the Nav1.7 interactome: Opportunities for chronic pain therapeutics.
Biochem Pharmacol, 2019May, 163 (9-20).
Vetter I
et al.
NaV1.7 as a pain target - From gene to pharmacology.
Pharmacol Ther, 2017Apr, 172 (73-100).
Eagles DA
et al.
Fifteen years of NaV 1.7 channels as an analgesic target: Why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy?
Br J Pharmacol, 2022Jul, 179 (3592-3611).
Rush AM
et al.
Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia.
J. Physiol. (Lond.),
1998
Sep
15
, 511 ( Pt 3) (771-89).
Contributors: Katherine Johnston
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/126/ , accessed on 2026 Jul 31