Nav1.4
Description: sodium channel, voltage-gated, type IV, alpha subunit Gene: Scn4a Alias: nav1.4, scn4a
Nav1.4, encoded by the gene scn4a, is a sodium, voltage-gated, type 4, alpha subunit channel. Nav1.4 is predominantly expressed in the CNS. It is primarily expressed in skeletal muscle cells and is responsible for the initiation of action potential in those tissues. Mutation in the channel are the cause of pathological enhanced or reduced muscle excitability
Experimental data
Mouse Nav1.4 gene in CHO host cells |
||
|
Click for details 
15 °Cshow 59 cells |
Click for details 
25 °Cshow 60 cells |
Click for details 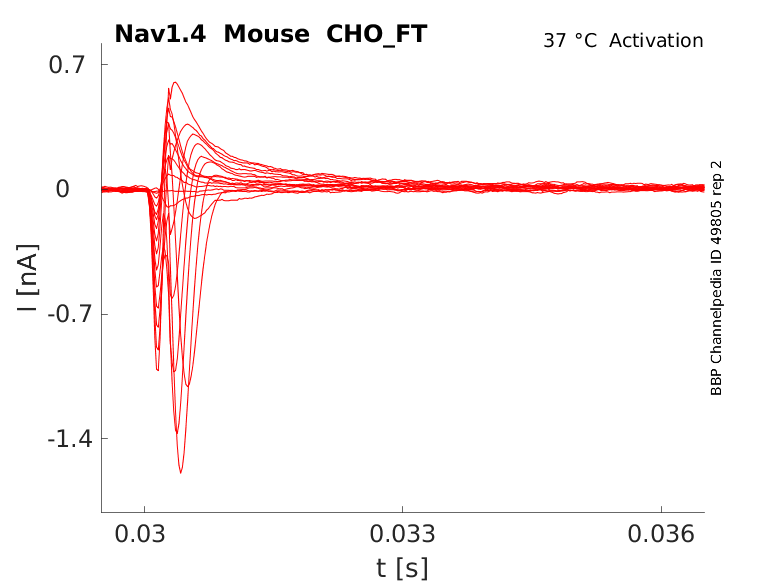
35 °Cshow 54 cells |
scn4a, the gene encoding for Nav1.4, is located on chromosome 17 at position 23-25 (17q23-25) in humans and is composed of 24 coding exons.
No alternative splicing events have been reported in humans but some scn4a variants have been identified in other species (rats, mouse, zebrafish) [2140] [2141].
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_000334.4 | 7805 | |
| Mouse | NM_133199.2 | 6598 | |
| Rat | NM_013178.2 | 6959 |
The human Nav1.4 protein is composed of 1836 amino acids with a molecular weight of around 227 Kda [2140]. [2140].
To date, there has been little research on the function characterisation of Nav1.4 isoforms
Isoforms
Like most mammalian proteins, Nav1.4 is subject to a series of post translational modifications (PTM).
Glycosylation can have a large impact on the gating properties of Nav1.4. N-linked glycans and O-linked glycans are often terminated by sialic acids, whose negative charge has been shown to shift the voltage dependence of activation and inactivation toward hyperpolarized potentials. This results in enhanced rates of fast inactivation, and reduced rates of recovery from fast inactivation [2142]
Phosphorylation can also induce a negative shift in the voltage dependence of inactivation for Nav1.4. Phosphorylation by PKC was shown to reduce peak Na+ currents by approximately 90%. This resulted in a -15 mV shift in the midpoint of steady-state inactivation and caused a slight speeding of inactivation [2143]
Reactive oxygen species (ROS) have also been implicated in the regulation of Nav1.4 activity. For example, Chloramine-T is a specific oxidant to sulphur-containing residues and can alter protein function through the oxidation of methionine residues. Chloramine-T has been found to remove more than 50% of Nav1.4 inactivation, with negligible impact on current activation [2144].
As Nav1.4 does not contain a PY motif, it is not targeted for ubiquitination. Instead, degradation is thought to occur via an alternative process that has yet to be elucidated [2145].
Visual Representation of Nav1.4 Structure
Methodology for visual representation of structure available here
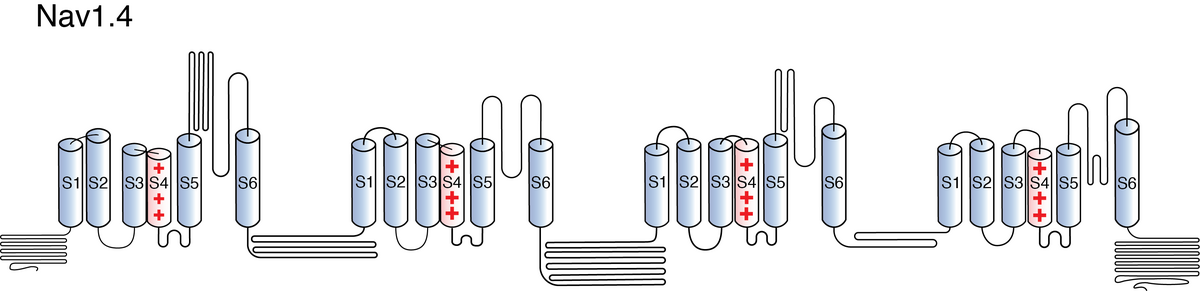
Like all voltage gated sodium channels, Nav1.4 is made up of a single protein comprised of 4 homologous domains (DI-DIV). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.4 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus deactivating the channel. Nav1.4 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of human Nav1.4, in complex with Navβ1, was resolved via cryo-electron microscopy, giving us a detailed insight to the structural features of Nav1.4. Structural resolution of the ion channel highlighted certain features such as an asymmetric selectivity filter (narrowest part of the open channel pore) and an up conformation for voltage sensing domains 1-4, as well as confirmed the structural basis for fast inactivation [2146].
The approximative size/surface of Nav1.4 can be determined via the resolved or predicted structures.
Nav1.4 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
The electrophysiological properties of Nav1.4 are similar to those of the CNS channels (Nav 1.1, 1.2, 1.3, 1.6) [881].
Nav1.4 activates rapidly in response to depolarization and conducts a large inward Na+ current, driving the rapid upstroke of the action potential [2147]. Fast inactivation quickly kicks in following the channel opening, with the protein returning to its resting state after repolarization.
Like other voltage gated sodium channels, upon prolonged depolarization, Nav1.4 channels progressively enter the slow inactivation (SI) state from which they are also slow to recover. In Nav1.4, this SI mechanisms are thought to be controlled by the immobilization of the voltage sensing domains following a prolonged depolarization [2148].
Single Channel Unitary Conductance
Single channel unitary conductance is determined experimentally.
For Nav1.4, there have been recordings of single channel unitary conductance values with some degree of variance between experiments:
- 24.9 pS - cRNA-injected Xenopus oocytes [2149]
- 52 pS - cRNA injected Xenopus oocytes [2150]
- 17.6 ± 0.2 pS - HEK [2151]
- 22 pS - HEK [2152]
Models
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Origin : HEK cells | Recording temperature: RT
Temperature dependence: Q10 | Formalism: Markov
States: C1, C2, O1, O2, I1, I2 | Implementation: NEURON
Simulation (Nav14_a.mod):

Membrane Systems Group 2022
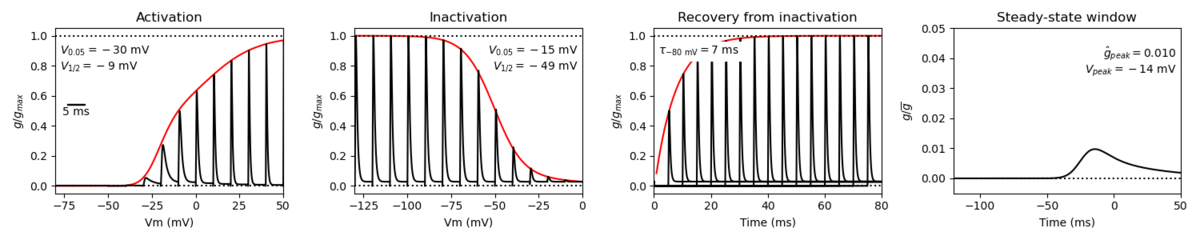
Origin : Mouse scn4a | Host cell: CHO
Recording temperature: 25 C | Formalism: Hodgkin-Huxley
Gates: m, h | Implementation: NEURON
Simulation (Nav14__mCHO25c.mod):
Tissue and cellular
Nav1.4 is predominantly expressed in skeletal muscles [2153] [2117].
Within skeletal muscles, Nav1.4 is expressed in the plasma membranes of both the transverse tubules and the sarcolemma [2147]. The distribution within these muscular tissues also varies, with the density of Nav1.4 being around 20 times higher at the neuromuscular junction (NMJ). This is a result of local mRNA accumulation, which ensures the proper transmission of the motoneuron action potential to the muscle fiber [2140] [2147].
Developmental
In rodents, scn4a expression increases just after birth [2140]. However, Nav1.4 expression is relatively low in rat neonatal skeleton muscles but highly expressed in adult rat skeletal muscles [881].
Expression modulation
Androgens may also play a role in the regulation of scn4a expression as mice with genes for modified androgens have been shown to have decreased scn4a expression levels. These mice also eventually die as a result of their neuro-muscular condition. Further research is needed to definitively confirm the role of androgens in scn4a expression and the exact molecular mechanisms of interaction [2158].
As Nav1.4 is predominantly expressed in muscle cells, their CNS sub-cellular distribution has not yet been characterized.
Muscle Action Potential Initiator
Given its location, Nav1.4 is responsible for almost all of the inward Na+ current that generates the action potential in skeletal muscles, leading to muscle contraction [2147].
Channelopathies
The crucial role of Nav1.4 in muscular action potential generation is highlighted by the numerous channelopathies that arise when the channel is deregulated. Mutations in Nav1.4 disrupt channel activity by impairing either fast and/or slow inactivation, or voltage-gating mechanism, resulting in pathologically enhanced or reduced muscle excitability. Known Nav1.4 channelopathies include [2154] [2147]:
- Congenital myasthenic syndrome
- Congenital myopathy with hypotonia
- Hyperkalaemic periodic paralysis
- Hypokalaemic periodic paralysis
- Normokalemic Periodic Paralysis
- Paramyotonia congenita
- Potassium-aggravated myotonia
- Sodium channel myotonia
- Rhabdomyolysis [2155]
Nav1.4 may interact with β subunits through specific binding sites. To date, evidence of only interactions with Navβ1 and Navβ4 has been identified.
[Nav1.4] [Navβ1] co-expression impacts both fast and slow inactivation compared to Nav1.4 expression alone. Fast inactivation is mildly disrupted with a 5 mV rightward shift. Following a 30 second depolarization, the voltage dependence of slow inactivation is shifted to the right (depolarized) and channel recovery is faster [2156].
[Nav1.4] [Navβ4] co-expression has been shown to lead to a modest negative shift in the voltage dependence of activation [406].
However, given the limited research on Nav1.4 and Navβ interactions, aside from Navβ1, further research is needed to determine the full extent of the role of Navβ on the channel.
There is clear evidence that the gating regulation and expression of Nav subunits relies on a variety of accessory and regulatory proteins. Despite this understanding, very little is known about proteins that interact with Nav1.4. However, some interactions, with Ankyrin-G, syntrophin, and Calmodulin (CaM), have been observed [2140].
Calmodulin is a small ubiquitous eukaryotic Ca2+-sensing regulatory protein. CaM can induce changes in target proteins via its binding to them and in response to changes in calcium concentration. Association with CaM is essential for the functional expression of Nav1.4. Disruptions of the interaction lead to a 99% reduction in Nav1.4 current amplitude [53]. Ca2+-sensing properties means that its activity can also be modulated based off of Ca2+ concentrations. Indeed, NaV1.4 exhibits a unique CaM mediated Ca2+-dependent inactivation (CDI). Under increased [Ca2+] levels, bulk Na+ current is reduced by approximately 30% [2157].
Nav1.4 channels are sensitive to nanomolar concentrations of tetrodotoxin (TTX), like the CNS channels. They are also sensitive to nanomolar concentrations of μ-conotoxin, which specifically interacts with Nav1.4 [881]
For additional resources on potential drug and compound interactions:
- Known and predicted drug interactions with Nav1.4
- Known and predicted animal toxin interactions with Nav1.4
References
Herzog RI
et al.
Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties.
J. Neurosci.,
2003
Sep
10
, 23 (8261-70).
Yu FH
et al.
Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2.
J. Neurosci.,
2003
Aug
20
, 23 (7577-85).
Catterall WA
et al.
International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels.
Pharmacol. Rev.,
2005
Dec
, 57 (397-409).
Goldin AL
Diversity of mammalian voltage-gated sodium channels.
Ann. N. Y. Acad. Sci.,
1999
Apr
30
, 868 (38-50).
Zhang J
et al.
N-type fast inactivation of a eukaryotic voltage-gated sodium channel.
Nat Commun, 20220517, 13 (2713).
Black JA
et al.
Noncanonical roles of voltage-gated sodium channels.
Neuron,
2013
Oct
16
, 80 (280-91).
Loussouarn G
et al.
Physiological and Pathophysiological Insights of Nav1.4 and Nav1.5 Comparison.
Front Pharmacol, 2015, 6 (314).
Liu H
et al.
A novel Na+ channel splice form contributes to the regulation of an androgen-dependent social signal.
J. Neurosci.,
2008
Sep
10
, 28 (9173-82).
Ednie AR
et al.
Sialic acids attached to N- and O-glycans within the Nav1.4 D1S5-S6 linker contribute to channel gating.
Biochim. Biophys. Acta,
2015
Feb
, 1850 (307-17).
Bendahhou S
et al.
Serine-1321-independent regulation of the mu 1 adult skeletal muscle Na+ channel by protein kinase C.
Proc. Natl. Acad. Sci. U.S.A.,
1995
Dec
19
, 92 (12003-7).
Kassmann M
et al.
Oxidation of multiple methionine residues impairs rapid sodium channel inactivation.
Pflugers Arch.,
2008
Sep
, 456 (1085-95).
Pei Z
et al.
Posttranslational Modification of Sodium Channels.
Handb Exp Pharmacol, 2018, 246 (101-124).
Pan X
et al.
Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1.
Science, 20181019, 362 ().
Silva JR
et al.
Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels I: wild-type skeletal muscle Na(V)1.4.
J. Gen. Physiol.,
2013
Mar
, 141 (309-21).
Chahine M
et al.
Functional expression and properties of the human skeletal muscle sodium channel.
Pflugers Arch.,
1994
May
, 427 (136-42).
Backx PH
et al.
Molecular localization of an ion-binding site within the pore of mammalian sodium channels.
Science,
1992
Jul
10
, 257 (248-51).
Carle T
et al.
Gating defects of a novel Na+ channel mutant causing hypokalemic periodic paralysis.
Biochem. Biophys. Res. Commun.,
2006
Sep
22
, 348 (653-61).
Simkin D
et al.
Mechanisms underlying a life-threatening skeletal muscle Na+ channel disorder.
J. Physiol. (Lond.),
2011
Jul
1
, 589 (3115-24).
Trimmer JS
et al.
Primary structure and functional expression of a mammalian skeletal muscle sodium channel.
Neuron,
1989
Jul
, 3 (33-49).
Mantegazza M
et al.
Sodium channelopathies of skeletal muscle and brain.
Physiol Rev, 20211001, 101 (1633-1689).
Vivante A
et al.
Exome sequencing in Jewish and Arab patients with rhabdomyolysis reveals single-gene etiology in 43% of cases.
Pediatr Nephrol, 2017Dec, 32 (2273-2282).
Webb J
et al.
Slow inactivation of the NaV1.4 sodium channel in mammalian cells is impeded by co-expression of the beta1 subunit.
Pflugers Arch.,
2009
Apr
, 457 (1253-63).
Ben-Johny M
et al.
Conservation of Ca(2+)/Calmodulin Regulation across Na and Ca(2+) Channels.
Cell,
2014
Jun
19
, 157 (1657-70).
Yu Z
et al.
Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model.
J. Clin. Invest.,
2006
Oct
, 116 (2663-72).
Contributors: Katherine Johnston
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/123/ , accessed on 2026 Aug 07