Kv7.2
Description: potassium voltage-gated channel, KQT-like subfamily, member 2 Gene: Kcnq2 Alias: KV7.2, EBN, BFNC, EBN1, ENB1, HNSPC, KCNA11, KVEBN1, KCNQ2
Kv7.2, encoded by KCNQ2, is a members of the potassium voltage-gated channel KQT-like subfamily.
Kv7.2 is also known as: KCNQ2; EBN; BFNC; EBN1; ENB1; BFNS1; EIEE7; HNSPC; KCNA11; KVEBN1.
Kv7.2 assembles with Kv7.3 to form the M channel, a slowly activating and deactivating potassium channel that plays a critical role in the regulation of neuronal excitability. Defects in this gene are a cause of benign familial neonatal convulsions type 1 (BFNC), also known as epilepsy, benign neonatal type 1 (EBN1) NCBI [690].
From an evolutionary perspective, KCNQ2 and KCNQ3 were the last KCNQ subunits to emerge, coincident with the apparition of myelin [701].
At least five transcript variants encoding five different isoforms have been found for this gene. [464]
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_172107.4 | 9247 | |
| Mouse | NM_010611.3 | 8301 | |
| Rat | NM_133322.2 | 4158 |
At least five transcript variants encoding five different isoforms have been found for this gene. [464]
Isoforms
Post-Translational Modifications
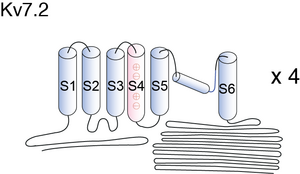
Visual Representation of Kv7.2 Structure
Methodology for visual representation of structure available here
STRUCTURE & MUTATION OF Kv7.2

Basic structure of KCNQ2/3 proteins and mutations leading to BFNC. KCNQ proteins have six transmembrane domains (TMDs) and a pore-forming P-loop. Mutations found in KCNQ2 (blue circles): Y284C, A306T, fs283, fs494 and spl516, spl397, fs534, fs616 and fs838. Numbering of KCNQ2 residues is according to KCNQ3 mutations (green squares). Several mutations truncate the channel before or in the A domain or the putative assembly domain, shown as beige and white boxes respectively. KCNQ4 mutations (red squares) identified in people with progressive dominant hearing loss DFNA2. With the exception of Fs71, these mutants exert dominant-negative effects. The L281S mutation73 has not been tested functionally. The equivalent tryptophan residue is mutated in KCNQ3 (W309R) and KCNQ4 (W276S), as well as in KCNQ1 in JLNS (W305S)74, indicating that it has no strong dominant-negative effect. G285 is the first glycine of the GYG pore signature sequence, which is also mutated in KCNQ1 in the dominant long-QT syndrome (G314S)75and suppresses wild-type KCNQ1 currents. Fs, frameshift mutation; spl, splice site mutation. Both types of mutations are expected to truncate the protein. c | Dendrogram of KCNQ and selected Kv channels [464]
Like all Kv channels, the KCNQ α subunits share a common core structure of six transmembrane segments with a voltage sensing domain (S1–S4) and a pore domain (S5 and S6)[696]. Sequence analysis predicts the presence of four helical regions (A–D) in all family members [697], and helices A and B constitute the binding site for calmodulin (CaM). [693] See figure 1 in [464] for the basic structure of KCNQ2/3 channels.
Mutation of the putative Gly hinge to Ala in KCNQ2 (Kv7.2) stops channel function.[77]
Kv7.2 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
KCNQ2/KCNQ3
KCNQ2/KCNQ3 heteromers yield currents with the properties of the M-current, see figure 2 in Jentsch's review [464].
Single Channel Kv7.2 Currents in CHO cells

KCNQ2/KCNQ3
KCNQ2 and KCNQ3 were identified by homology to KCNQ1, and also by positional cloning in families with benign familial neonatal convulsions (BFNC), a neonatal form of epilepsy. Both subunits are expressed mainly in neuronal tissue including sympathetic ganglia, and their expression patterns in the brain overlap extensively. However, in situ hybridization indicates that they are not always expressed in the same ratio, and immunocytochemistry has shown that some neurons stain only for one or the other subunit. [464]
M-type channels are generated by the KCNQ (Kv7) family of voltage-gated subtypes [695], and they are found throughout the nervous system where they fulfil dominant roles in the control of excitability and neural discharges [464]. The M channel is slowly activating and deactivating potassium conductance important for determining the subthreshold electroexcitability in the central nervous system, especially in neocortical, thalamic, and hippocampal neurons. Therefore, heteromers of KCNQ2 with KCNQ3 or KCNQ5 forming M-type current potassium channels play a crucial role in the modulation of neuronal excitability. [694]
KCNQ2 and KCNQ3 are coexpressed on the cell bodies and dendrites of many hippocampal and cortical neurons. [461]
DISTRIBUTION OF KCNQ2 in NEURON

KCNQ2 plays a functional role at axonal initial segments and nodes of Ranvier.[339]
Benign Familial Neonatal Convulsions are a rare epilepsy disorder with an autosomal-dominant inheritance. It is linked to mutations in the potassium channel genes KCNQ2 and KCNQ3. These encode for Kv7.2 and Kv7.3. [692] KCNQ2 mutation has implications for diagnosis and prognosis of familial neonatal seizures. [691]
further, KCNQ2/3 play a role in idiopathic generalized epilepsies and Rolandic epilepsy. [694]
Calmodulin bound to KCNQ2 acts as a Ca2+ sensor, conferring Ca2+ dependence to the trafficking of the channel to the plasma membrane. [693]
Syntaxin 1A
Syntaxin 1A expressed with KCNQ2 homomeric channels resulted in a 2-fold reduction in macroscopic conductance and 2-fold slower activation kinetics.[58]
Extracellular H+ ions
Whole-cell and single-channel recordings demonstrated that extracellular H+ ions effect heterologously expressed KCNQ2/3 channels in the following way: KCNQ2/3 current was inhibited by H+ ions with an IC50 of 52 nM (pH 7.3) at -60 mV, rising to 2 microM (pH 5.7) at -10 mV. Neuronal M-current exhibited a similar sensitivity. I.e. extracellular H+ ions affected two distinct properties of KCNQ2/3 current: the maximum current attainable upon depolarization (Imax) and the voltage dependence of steady-state activation. [66]
Mepyramine and Diphenhydramine
Mepyramine and diphenhydramine, two structurally related first-generation antihistamines, can act as potent KCNQ/M channel blockers. Extracellular application of these drugs quickly and reversibly reduced KCNQ2/Q3 currents heterologously expressed in HEK293 cells. [72]
Meclofenamate and Diclofenac
Meclofenamic acid (meclofenamate) and diclofenac, two related molecules previously used as anti-inflammatory drugs, act as KCNQ2/Q3 channel openers. Extracellular application of meclofenamate (EC(50) = 25 microM) and diclofenac (EC(50) = 2.6 microM) resulted in the activation of KCNQ2/Q3 K(+) currents by causing a hyperpolarizing shift of the voltage activation curve and markedly slowing the deactivation kinetics. The effects of the drugs were stronger on KCNQ2 than on KCNQ3 channel alpha subunits but they did not enhance KCNQ1 K(+) currents. Both openers increased KCNQ2/Q3 current amplitude at physiologically relevant potentials and led to hyperpolarization of the resting membrane potential. [78]
CELECOXIB
Interestingly, celecoxib, a COX-2-specific inhibitor, has been shown to enhance Kv7.2–Kv7.5 currents overexpressed in HEK 293 cells with an EC50 of 2–5 µM. Previously, celecoxib had been shown to enhance Kv7.5 currents in A7r5 rat aortic smooth muscle cells and cause a vasodilatation of rat mesenteric arteries, whereas other COX-2-specific inhibitors, such as rofecoxib (Vioxx™; Merck & Co. Inc., Whitehouse Station, NJ, USA), had no effect on the currents [1675]
References
Selective interaction of syntaxin 1A with KCNQ2: possible implications for specific modulation of presynaptic activity.
PLoS ONE, 2009 , 4 (e6586).
Xiong Q
et al.
Combinatorial augmentation of voltage-gated KCNQ potassium channels by chemical openers.
Proc. Natl. Acad. Sci. U.S.A.,
2008
Feb
26
, 105 (3128-33).
Prole DL
et al.
Mechanisms underlying modulation of neuronal KCNQ2/KCNQ3 potassium channels by extracellular protons.
J. Gen. Physiol.,
2003
Dec
, 122 (775-93).
Fedorenko O
et al.
A schizophrenia-linked mutation in PIP5K2A fails to activate neuronal M channels.
Psychopharmacology (Berl.),
2008
Jul
, 199 (47-54).
Liu B
et al.
Antihistamine mepyramine directly inhibits KCNQ/M channel and depolarizes rat superior cervical ganglion neurons.
Neuropharmacology,
2008
Mar
, 54 (629-39).
Zaika O
et al.
Determinants within the turret and pore-loop domains of KCNQ3 K+ channels governing functional activity.
Biophys. J.,
2008
Dec
, 95 (5121-37).
Miceli F
et al.
Gating consequences of charge neutralization of arginine residues in the S4 segment of K(v)7.2, an epilepsy-linked K+ channel subunit.
Biophys. J.,
2008
Sep
, 95 (2254-64).
Maljevic S
et al.
Nervous system KV7 disorders: breakdown of a subthreshold brake.
J. Physiol. (Lond.),
2008
Apr
1
, 586 (1791-801).
Soldovieri MV
et al.
Atypical gating of M-type potassium channels conferred by mutations in uncharged residues in the S4 region of KCNQ2 causing benign familial neonatal convulsions.
J. Neurosci.,
2007
May
2
, 27 (4919-28).
Seebohm G
et al.
Differential roles of S6 domain hinges in the gating of KCNQ potassium channels.
Biophys. J.,
2006
Mar
15
, 90 (2235-44).
Peretz A
et al.
Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties.
Mol. Pharmacol.,
2005
Apr
, 67 (1053-66).
Wuttke TV
et al.
Neutralization of a negative charge in the S1-S2 region of the KV7.2 (KCNQ2) channel affects voltage-dependent activation in neonatal epilepsy.
J. Physiol. (Lond.),
2008
Jan
15
, 586 (545-55).
Bentzen BH
et al.
The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels.
Neuropharmacology,
2006
Nov
, 51 (1068-77).
Bibbig A
et al.
Self-organized synaptic plasticity contributes to the shaping of gamma and beta oscillations in vitro.
J. Neurosci.,
2001
Nov
15
, 21 (9053-67).
Traub RD
et al.
Fast rhythmic bursting can be induced in layer 2/3 cortical neurons by enhancing persistent Na+ conductance or by blocking BK channels.
J. Neurophysiol.,
2003
Feb
, 89 (909-21).
Migliore M
et al.
Computer simulations of morphologically reconstructed CA3 hippocampal neurons.
J. Neurophysiol.,
1995
Mar
, 73 (1157-68).
Dedek K
et al.
Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel.
Proc. Natl. Acad. Sci. U.S.A.,
2001
Oct
9
, 98 (12272-7).
Jentsch TJ
Neuronal KCNQ potassium channels: physiology and role in disease.
Nat. Rev. Neurosci.,
2000
Oct
, 1 (21-30).
Yum MS
et al.
The first Korean case of KCNQ2 mutation in a family with benign familial neonatal convulsions.
J. Korean Med. Sci.,
2010
Feb
, 25 (324-6).
Goldberg-Stern H
et al.
Novel mutation in KCNQ2 causing benign familial neonatal seizures.
Pediatr. Neurol.,
2009
Nov
, 41 (367-70).
Volkers L
et al.
Functional analysis of novel KCNQ2 mutations found in patients with Benign Familial Neonatal Convulsions.
Neurosci. Lett.,
2009
Oct
2
, 462 (24-9).
Alaimo A
et al.
Calmodulin activation limits the rate of KCNQ2 K+ channel exit from the endoplasmic reticulum.
J. Biol. Chem.,
2009
Jul
31
, 284 (20668-75).
Hahn A
et al.
Sodium and potassium channel dysfunctions in rare and common idiopathic epilepsy syndromes.
Brain Dev.,
2009
Aug
, 31 (515-20).
Wang HS
et al.
KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel.
Science,
1998
Dec
4
, 282 (1890-3).
Haitin Y
et al.
The C-terminus of Kv7 channels: a multifunctional module.
J. Physiol. (Lond.),
2008
Apr
1
, 586 (1803-10).
Yus-Najera E
et al.
The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels.
J. Biol. Chem.,
2002
Aug
9
, 277 (28545-53).
Hill AS
et al.
Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates.
PLoS Genet.,
2008
Dec
, 4 (e1000317).
Jepps TA
et al.
One man's side effect is another man's therapeutic opportunity: targeting Kv7 channels in smooth muscle disorders.
Br. J. Pharmacol.,
2013
Jan
, 168 (19-27).
Cooper EC
et al.
Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy.
Proc. Natl. Acad. Sci. U.S.A.,
2000
Apr
25
, 97 (4914-9).
Li Y
et al.
Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate.
J. Neurosci.,
2005
Oct
26
, 25 (9825-35).
Miceli F
et al.
Early-Onset Epileptic Encephalopathy Caused by Gain-of-Function Mutations in the Voltage Sensor of Kv7.2 and Kv7.3 Potassium Channel Subunits.
J. Neurosci.,
2015
Mar
4
, 35 (3782-93).
Contributors: Rajnish Ranjan, Michael Schartner, Nitin Khanna, Katherine Johnston
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/24/ , accessed on 2026 Aug 02