Kv8.2
Description: potassium channel, subfamily V, member 2 Gene: Kcnv2 Alias: Kv8.2, kcnv2, RCD3B
Kv8.2 (also known as RCD3B; KV11.1; MGC120515), encoded by the gene KCNV1, is member 2 of subfamily V. Kv8.2 is identified as a 'silent subunit', and it does not form homomultimers, but forms heteromultimers with several other subfamily members. Through obligatory heteromerization, it exerts a function-altering effect on other potassium channel subunits. This protein is strongly expressed in pancreas and has a weaker expression in several other tissues. [730] NCBI
Experimental data
Rat Kv8.2 gene in CHO host cells datasheet |
||
|
Click for details 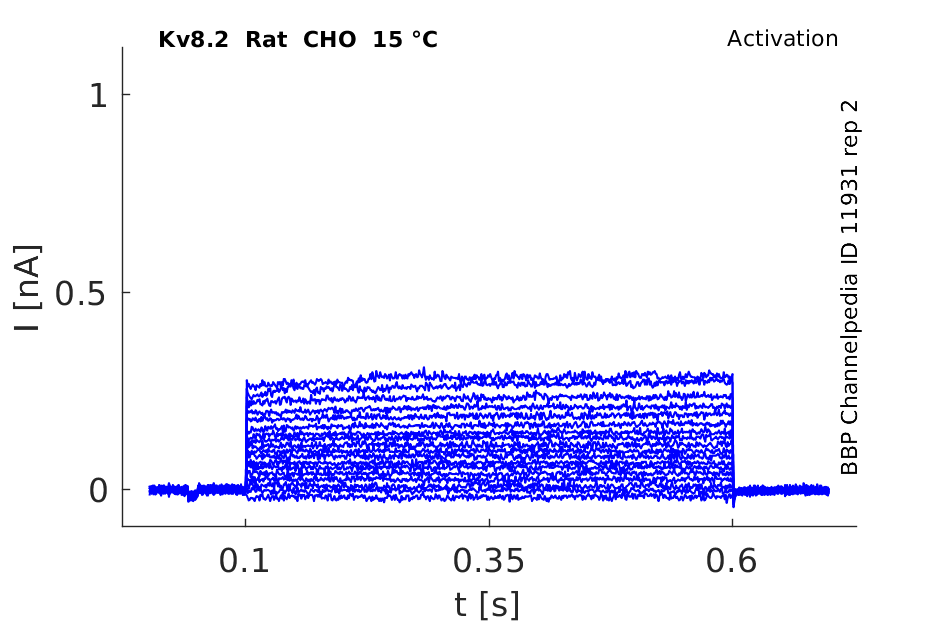
15 °Cshow 64 cells |
Click for details 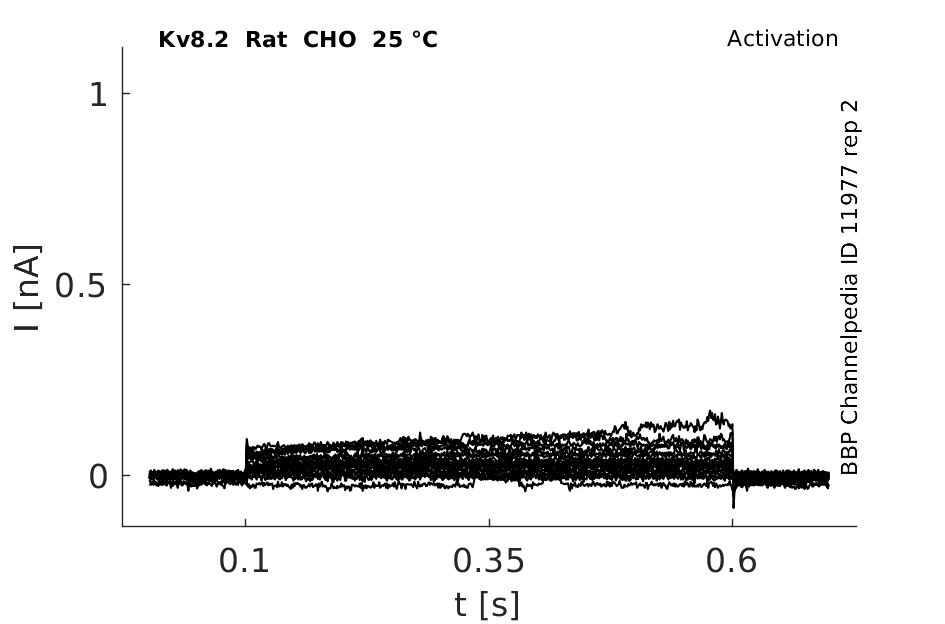
25 °Cshow 52 cells |
Click for details 
35 °Cshow 80 cells |
Gene
Transcript
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_133497.4 | 2178 | |
| Mouse | NM_183179.1 | 5092 | |
| Rat | NM_001106370.1 | 5201 |
Protein Isoforms
Isoforms
Post-Translational Modifications
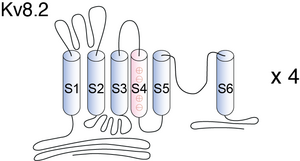
Visual Representation of Kv8.2 Structure
Methodology for visual representation of structure available here
Analysis of Mutations Located in the Pore Domain Kv8.2
The following figure shows the location of the disease-causing mutations in hKv8.2 in CDSRE patients examined in this study. Three of these, W450G, G459D, and G461R, are located in the pore region of the hKv8.2 α-subunit. The missense mutations G459D and G461R affect the first and second glycine, respectively, of the Gly-Tyr-Gly motif, the characteristic potassium channel signature sequence. To understand the functional consequences of these mutations, the corresponding mutations (W467G, G476D, and G478R) were introduced into mKv8.2, and their effect on subunit localization in COS7L cells was examined. Like mKv8.2, the expression of either mKv8.2-W467G-EGFP, mKv8.2-G476D-EGFP, or mKv8.2-G478R-EGFP resulted in an intracellular localization [1753]
Voltage-gated K+ channels selectively transfer potassium ions through the plasma membrane in response to depolarization. The ion-conducting core of voltage-gated K+ channels is composed of four Kv-alpha subunits, which also possess the voltage sensor. According to structural similarities, members of the Kv family were initially divided into four subfamilies (Kv1–Kv4), the mammalian equivalents of the Drosophila Shaker, Shab, Shaw, and Shal gene products, respectively. These Kv subunits give rise to functional homotetrameric K+ channels, when they are expressed heterologously in Xenopus laevis oocytes (for review see [496]). Coexpression of members of the same subfamily may result in functional heterotetrameric channels and currents with properties different from those of the homotetramers of the parent subunits. The current of these heteromeric Kv channels was also identified in native tissues [733], [734]. Subunits belonging to different Kv1–Kv4 subfamilies do not coassemble [731]. The selectivity within the subfamilies is ensured by the specific N-terminal tetramerization domains [598], [599].
Kv8.2 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Mutated (B6-Kv8.2) Kinetics whit Kv2.1 in CHO cells

hKv8.2 and hKv2.1 Co transfected in CHO Cell Kinetics

Kv8.2 Expressed in the Brain
Members of the Kv2 family are expressed in the nervous system and underlie the neuronal delayed rectifier K+ current, which is important for limiting membrane excitability, particularly under conditions of repetitive stimulation [91]. In the hippocampus, which is a region of particular importance for seizure generation, Kv2.1 is the major contributor to the delayed rectifier potassium current [732] and colocalizes with Kv8.2 in hippocampal pyramidal neurons (Allen Institute for Brain Science (2009) Allen mouse brain atlas. Available at: http:// mouse.brain-map.org. Accessed August 13, 2010.)
Whole-brain expression of Kcnv2 does not differ between strains B6 and SJL. We further investigated strain-dependent expression in the hippocampus for three reasons: first, previous EEG studies showed that seizures in Scn2aQ54 likely originate in the hippocampus; second, Kcnv2 transcripts are enriched in the hippocampus; and, third, the biophysical differences described above were counterintuitive and suggested more complex physiology. RNA was isolated from dissected hippocampi of 8-wk-old B6 and SJL mice for quantitative RT-PCR (qRT-PCR) analysis [730]
Membrane Translocation
Kv8.2 can form functional heterotetramers with Kv2 subunits and influence membrane translocation and channel properties [648], [731].
Kcnv2 transgene-mediated transfer of seizure susceptibility is observed in mice, as well as human variants that alter delayed rectifier K+ currents. These results identify KCNV2 as an epilepsy gene in mice and humans. [730]
Cone Dystrophy
Mutations in KCNV2 have been proposed as the molecular basis for cone dystrophy with supernormal rod electroretinogram. KCNV2 codes for the modulatory voltage-gated potassium channel α-subunit, Kv8.2, which is incapable of forming functional channels on its own. Functional heteromeric channels are however formed with Kv2.1 in heterologous expression systems, with both α-subunit genes expressed in rod and cone photoreceptors [1753]
Epilepsy
Kv8.2 also contributes to Epilepsy [730]
Kv2.1
Kv8.2 changes the kinetics of Kv2.1 (see kinetics for more info) [730]
References
Misonou H
et al.
Kv2.1: a voltage-gated k+ channel critical to dynamic control of neuronal excitability.
Neurotoxicology,
2005
Oct
, 26 (743-52).
Coetzee WA
et al.
Molecular diversity of K+ channels.
Ann. N. Y. Acad. Sci.,
1999
Apr
30
, 868 (233-85).
Li M
et al.
Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel.
Science,
1992
Aug
28
, 257 (1225-30).
Shen NV
et al.
Molecular recognition and assembly sequences involved in the subfamily-specific assembly of voltage-gated K+ channel subunit proteins.
Neuron,
1995
Mar
, 14 (625-33).
Ottschytsch N
et al.
Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome.
Proc. Natl. Acad. Sci. U.S.A.,
2002
Jun
11
, 99 (7986-91).
Jorge BS
et al.
Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility.
,
2011
Mar
14
, ().
Czirják G
et al.
Characterization of the heteromeric potassium channel formed by kv2.1 and the retinal subunit kv8.2 in Xenopus oocytes.
J. Neurophysiol.,
2007
Sep
, 98 (1213-22).
Murakoshi H
et al.
Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons.
J. Neurosci.,
1999
Mar
1
, 19 (1728-35).
Sheng M
et al.
Presynaptic A-current based on heteromultimeric K+ channels detected in vivo.
Nature,
1993
Sep
2
, 365 (72-5).
Wang H
et al.
Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons.
Nature,
1993
Sep
2
, 365 (75-9).
Smith KE
et al.
Functional analysis of missense mutations in Kv8.2 causing cone dystrophy with supernormal rod electroretinogram.
J. Biol. Chem.,
2012
Dec
21
, 287 (43972-83).
Wu H
et al.
Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause "cone dystrophy with supernormal rod electroretinogram" in humans.
Am. J. Hum. Genet.,
2006
Sep
, 79 (574-9).
Contributors: Rajnish Ranjan, Michael Schartner, Katherine Johnston
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/29/ , accessed on 2026 Aug 04