Nav1.6
Description: sodium channel, voltage gated, type VIII, alpha subunit Gene: Scn8a Alias: nav1.6, scn8a
Nav1.6, encoded by the gene scn8a, is a sodium, voltage-gated, type 8, alpha subunit channel. Nav1.6 is most abundantly expressed in the CNS during adulthood. It is involved in the control of backpropagation and peaking of the action potential. Mutations to the channel are linked to developmental and epileptic encephalopathy .
Experimental data
Mouse Nav1.6 gene in CHO host cells |
||
|
Click for details 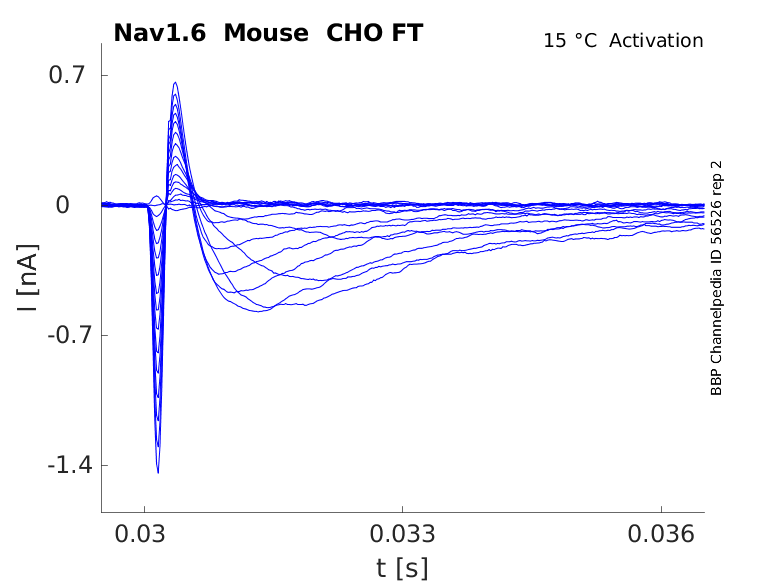
15 °Cshow 52 cells |
Click for details 
25 °Cshow 58 cells |
Click for details 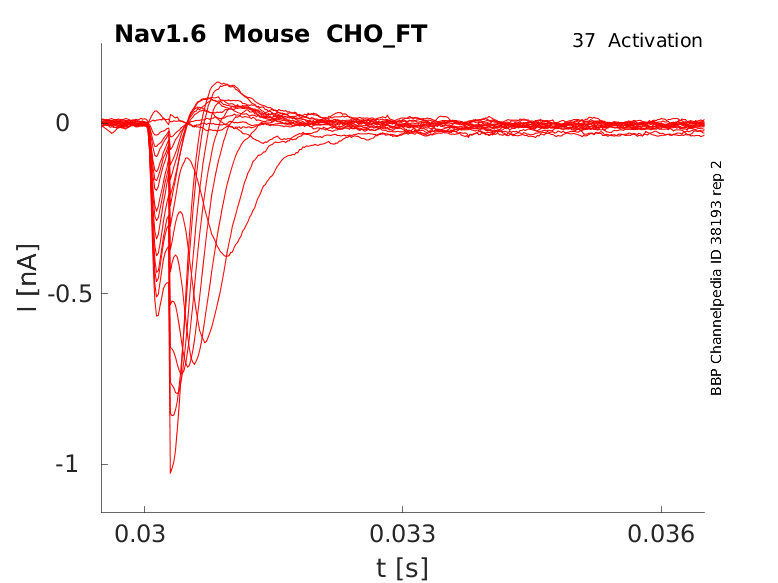
35 °Cshow 60 cells |
scn8a, the gene encoding for Nav1.6, is located on chromosome 12 at position 13 (12q13.13) in humans and is composed of 27 exons 26 of which are coding and exon 1 being non-coding.
The amino acid sequence of Nav1.6 is 84% identical to the sequences of Nav1.1 and Nav1.2, but it is more distant from those two than they are to each other [52]
There exists multiple Nav1.6 transcript variants across species as a result of the alternative splicing of scn8a (see Protein Isoform table).
Of the many variants that have been identified, only a few have been properly studied, such the ones resulting from the alternative splicing of exons 10B, 12, 18, and 5.
Notably, like other Navs, the alternative splicing of exon 5 results in a “neonatal” variant of Nav1.6. This splicing event is mirrored by the alternative splicing of exon 18, leading to exon18A and exon18N variants. Exons 5 and 18 encode the corresponding transmembrane segments in domains I and III respectively, indicating possible shared evolutionary origin. Both variants are developmentally regulated, with their expression being highest during the fetal development stage before switching to the adult splice form. [2184] [21845] [2186]
mRNA transcripts, including exon 18N, are expressed at a low level in non-neuronal tissues and are subject to nonsense-mediated decay. 18A variants appear to be restricted to neurons, and are mediated by neuron-specific splice factors. The reasons for this control haven’t been determined yet but a hypothesis is that exon 18 may represent a fail-safe mechanism to prevent damage to non-neuronal cells [2186].
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_014191.4 | 11559 | |
| Mouse | NM_001077499.2 | 11340 | |
| Rat | NM_019266.3 | 6548 |
The canonical Nav1.6 isoform is made of around 1980 amino acid residues, the sequence of which is highly conserved across species. The channel has a molecular weight of 229 Kda. [2184]
There exists a number of protein isoforms that arise from the translation of the aforementioned transcript variants:
- The neonatal isoform, resulting from the alternative splicing of exon 5, differs by a single amino acid from the canonical adult form 5A.
- Proteins resulting from the translation of the exon 18N variant contain an in-frame stop codon that results in a truncated channel protein. [2186]
Isoforms
Glycosylation is a particularly important PTM as it is crucial for the biosynthesis, folding, and trafficking of voltage gated sodium channels. Glycosylation plays an important role in the proper subcellular localization of Nav1.6. Mice with aberrant glycosylation sites have reduced channel activity and defective neuronal localization, which manifested itself as chronic movement disorders. [21867][2188]
Nav1.6 appears to be relatively resistant to the modulation by kinases that are classically involved in the phosphorylation of neuronal channels, such as PKA and PKC. However, the channel is still subject to phosphorylation by a number of other enzymes. These include CamKII, p38 mitogen-activated protein kinase (MAPK), and glycogen synthase kinase-3 (GSK3). CaMKII and GCK3 both play a role in the regulation of excitability, as inhibition of the enzymes lead to a decrease in transient and persistent Nav1.6 sodium current and a depolarising shift in the voltage dependence of activation. MAPK is also indirectly involved in neuronal excitability by modulating the surface expression of the channel. Indeed p38 MAPK phosphorylation of Nav1.6 promotes Nedd4-ubiquitination, leading to increased degradation of the protein and thus decreased sodium current. [2189][2188]
Ubiquitination is responsible for the internalization and degradation of proteins, a process mediated by ubiquitin ligases which bind to PY motifs. Nav1.6 contains several PY motifs and undergoes ubiquitin-dependent modulation by Nedd4-a. Abrogation of Nedd4 leads to decreased Nav.6 internalization and an increase in current. However, the activity of Nedd4 is promoted by the previously mentioned p38 MAPK, indicating potential complex interactions between different PTMs, that modulate channel physiology and excitability. [2188]
Palmitoylation is involved in multiple stages of the life cycle of ion channels. Nav1.6 contains multiple identified palmitoylation sites, whose binding leads to the modulation of voltage-dependence of inactivation and increases in current density. Removal of these sites results in a reduction in Nav1.6 excitability [2188][2190].
Visual Representation of Nav1.6 Structure
Methodology for visual representation of structure available here

Like all voltage gated sodium channels, Nav1.6 is made up of a single protein comprised of 4 homologous domains (DI-DIV). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.6 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus deactivating the channel. Nav1.6 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of rat Nav1.6 was resolved via cryo-electron microscopy, giving us a detailed insight into the channel’s architecture. Structural resolution of the ion channel highlighted certain features such as the structural variations of the voltage sensing domain IV, which has a wider pocket due to the displacement of its S1–S3. VSDIV represents a major target for most Nav inhibitors of high affinity and selectivity. [2191] However, it is worth noting that the aforementioned study was done on Nav1.6 bound to other molecules and the protein was not in an active conformation state. Further research is needed to confirm the results of the structure of Nav1.6 alone in and active state
Nav1.6 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Nav1.6 displays similar kinetics to other voltage-gated sodium channels, namely fast activation and fast inactivation kinetics. However, it does have its own distinct current properties. Nav1.6 has a hyperpolarized shift in voltage-dependent activation compared to other Navs [2188]. Nav1.6 also has a more hyperpolarized voltage-dependence of steady-state inactivation, faster development of closed-state inactivation, slower kinetics of open-channel inactivation and a greater propensity to generate persistent and resurgent currents.[2192]
Single channel unitary conductance
Single channel unitary conductance is determined experimentally.
For Nav1.6, single channel unitary conductance has yet to be determined experimentally.
Model
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Origin : HEK293 cells | Recording temperature: RT
Temperature dependence: Q10 | Formalism: Markov
States: C1, C2, O1, O2, I1, I2 | Implementation: NEURON
Simulation (Nav16_a.mod):

Membrane Systems Group 2022
Origin : Mouse scn8a | Host cell: CHO
Recording temperature: 25 C | Formalism: Hodgkin-Huxley
Gates: m, h | Implementation: NEURON
Simulation (Nav16__mCHO25c__0106.mod):

Model Nav1.6 (ID=33)
| Animal | rat | |
| CellType | L5PC | |
| Age | 21 Days | |
| Temperature | 23.0°C | |
| Reversal | 50.0 mV | |
| Ion | Na + | |
| Ligand ion | ||
| Reference | [288] A L Goldin et. al; J. Neurosci. 1998 Aug 15 | |
| mpower | 1.0 | |
| m Inf | 1.0000/(1+ exp(-0.03937*4.2*(v - -17.000))) | |
| m Tau | 1 | |

Tissue & cellular
Nav1.6 is broadly expressed in the central nervous system (CNS) where it is predominantly expressed in a variety of excitatory and inhibitory neuronal cell types. These include the hippocampal pyramidal and granule cells, retinal ganglion cells, cortical pyramidal neurons, motor neurons, and cerebellar Purkinje and granule cells.
Nav1.6 is also expressed, at lower levels, in the peripheral nervous system (PNS) and is present in a variety of ganglion cells, such as the dorsal root ganglion and trigeminal ganglion neurons, where it is plays a crucial role in peripheral sensory neuron transduction. Nav1.6 is also found at a low level in cardiomyocytes [2188].
Nav1.6 has been identified in a number non-canonical, non-excitable cells where they are thought to play non-negligeable roles. For example, microglia have been shown to express Nav1.6 and removal of the channel lead to a 65% decrease in mice microglia phagocytic capacity of microglia lacking Nav1.6 [2117].
Developmental
Nav1.6 can be found at low levels during prenatal development before its expression levels increase postnatally [365][2193]
Within the neurons of the CNS, Nav1.6 is found mainly in the distal end of the axon initial segment (AIS), around 25–50 μm from the soma [2194]. It is also present at the nodes of Ranvier within myelinated neurons [2195].
Given its fast kinetics, Nav1.6 is involved in the upstroke of the action potential. However, Nav1.6 is also responsible for persistent and resurgent currents.
Navs produce a noncanonical type of non-inactivating sodium current, known as persistent current. This phenomenon is 5-fold higher in Nav1.6 [2196]. Though these currents are generally small, the summation of persistent sodium currents from multiple channels can amplify subthreshold neuronal inputs. This leads to the lowering of AP initiation threshold and enables repetitive AP firing. [2188].
Resurgent current is a voltage and time-dependent property of Nav1.6 which occurs after depolarization at intermediate repolarizing potentials and elicits a small, transient current. These contribute to the spontaneous firing and multi-peaked AP[2188].
Given the location and high channel density at the AIS, Nav1.6’s fast kinetics and persistent and resurgent current properties make it responsible for the initiation of AP as well as repetitive firing of neurons [2194][2197]
Channelopathies
Given its role in action potential initiation, changes in Nav1.6 channel excitability lead to a number of pathologies, the vast majority of which are linked to severe developmental and epileptic encephalopathy (DEE) [2198]
The type of pathology often depends on the type of mutation that occurs [2199]:
- Loss of function: Intellectual disability, movement disorder, ataxia
- Mild gain of function: benign infantile onset seizures, paroxysmal dyskinesia with normal cognition
- Moderate gain of function: Moderate epileptic encephalopathy, developmental arrest and regression
- Severe gain of function: developmental delay. early onset refractory seizures, non ambulatory. lack of speech and language, cortical visual impairement
Nav1.6 also presents high expression in various metastatic tumors, including cancers of the breast, prostate, lymph node, and cervix, and is believed to contribute toward cancer metastasis [2188]
Nav1.6, like other voltage-gated sodium channels, is subject to extensive regulation by various auxiliary proteins and secondary messengers. These regulatory proteins play an important role in the development, localization, and expression of the channel protein.
The binding of Nav β subunits to Nav1.6 regulates neuronal function of the channel. Navβ1 was shown to be necessary for proper localization of Nav1.6 to the AIS during development [2200] Navβ4 is responsible for increased neuronal excitability as a result of increased resurgent current when bound to Nav1.6 [2201] However, expression of Navβ4 and Nav1.6 alone, in a heterologous system, was not sufficient to produce increases in sodium current, suggesting that interactions with other proteins are necessary to mediate this phenomenon [2188]
The activity of fibroblast growth factor homologous factors (FHF) influence current density and gating properties of Nav1.6. FHF4B was shown to suppress Nav1.6 sodium current and regulate localisation to the AIS. FHF2A and FHF2B both impact the gating properties of the channel, resulting in changes in sodium current kinetics and changes in neuron excitability [2188]
Calmodulin (CaM) is a small ubiquitously expressed protein which binds/senses calcium ions. CaM affects several different properties of Nav1.6, including channel inactivation and persistent current. Disruption of Cam activity led to a 62% reduction in current amplitude. The interaction between Nav1.6 and CaM displays calcium dependent inactivation (CDI) as calmodulin was shown to be able to modulate the inactivation kinetics of Nav1.6 currents in a calcium-dependent manner [53]
Table of known and predicted drug interactions with Nav1.6
Table of known and predicted animal toxin interactions with Nav1.6
References
Lee A
et al.
Role of the terminal domains in sodium channel localization.
Channels (Austin),
2009 May-Jun
, 3 (171-80).
Royeck M
et al.
Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons.
J. Neurophysiol.,
2008
Oct
, 100 (2361-80).
Mercer JN
et al.
Nav1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons.
J. Neurosci.,
2007
Dec
5
, 27 (13552-66).
Shirahata E
et al.
Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1.6.
J. Neurophysiol.,
2006
Sep
, 96 (1347-57).
Rush AM
et al.
Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones.
J. Physiol. (Lond.),
2005
May
1
, 564 (803-15).
Zhou W
et al.
Use-dependent potentiation of the Nav1.6 sodium channel.
Biophys. J.,
2004
Dec
, 87 (3862-72).
Herzog RI
et al.
Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties.
J. Neurosci.,
2003
Sep
10
, 23 (8261-70).
Smith MR
et al.
Functional analysis of the mouse Scn8a sodium channel.
J. Neurosci.,
1998
Aug
15
, 18 (6093-102).
Lorincz A
et al.
Cell-type-dependent molecular composition of the axon initial segment.
J. Neurosci.,
2008
Dec
31
, 28 (14329-40).
Sittl R
et al.
Sustained increase in the excitability of myelinated peripheral axons to depolarizing current is mediated by Nav1.6.
,
2011
Feb
2
, ().
He H
et al.
Molecular determination of selectivity of the site 3 modulator (BmK I) to sodium channels in the CNS: a clue to the importance of Nav1.6 in BmK I-induced neuronal hyperexcitability.
Biochem. J.,
2010
Sep
28
, 431 (289-98).
Lorincz A
et al.
Molecular identity of dendritic voltage-gated sodium channels.
Science,
2010
May
14
, 328 (906-9).
Chatelier A
et al.
Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis.
Pflugers Arch.,
2010
Jun
, 460 (77-86).
Low SE
et al.
Na(v)1.6a is required for normal activation of motor circuits normally excited by tactile stimulation.
Dev Neurobiol,
2010
Jun
, 70 (508-22).
Osorio N
et al.
Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells.
J. Physiol. (Lond.),
2010
Feb
15
, 588 (651-70).
Duflocq A
et al.
Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments.
Mol. Cell. Neurosci.,
2008
Oct
, 39 (180-92).
Mechaly I
et al.
Molecular diversity of voltage-gated sodium channel alpha subunits expressed in neuronal and non-neuronal excitable cells.
Neuroscience,
2005
, 130 (389-96).
Grieco TM
et al.
Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin.
J. Neurosci.,
2004
Jan
7
, 24 (35-42).
Tzoumaka E
et al.
Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system.
J. Neurosci. Res.,
2000
Apr
1
, 60 (37-44).
Raman IM
et al.
Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice.
Neuron,
1997
Oct
, 19 (881-91).
Meeks JP
et al.
Action potential initiation and propagation in CA3 pyramidal axons.
J. Neurophysiol.,
2007
May
, 97 (3460-72).
Craner MJ
et al.
Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger.
Proc. Natl. Acad. Sci. U.S.A.,
2004
May
25
, 101 (8168-73).
Chabbert C
et al.
Voltage-gated Na+ channel activation induces both action potentials in utricular hair cells and brain-derived neurotrophic factor release in the rat utricle during a restricted period of development.
J. Physiol. (Lond.),
2003
Nov
15
, 553 (113-23).
Hossain WA
et al.
Where is the spike generator of the cochlear nerve? Voltage-gated sodium channels in the mouse cochlea.
J. Neurosci.,
2005
Jul
20
, 25 (6857-68).
Eijkelkamp N
et al.
Neurological perspectives on voltage-gated sodium channels.
Brain,
2012
Sep
, 135 (2585-612).
Kearney JA
et al.
Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6).
Hum. Mol. Genet.,
2002
Oct
15
, 11 (2765-75).
Saleh S
et al.
Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes.
J. Physiol. (Lond.),
2005
Oct
1
, 568 (155-69).
Trudeau MM
et al.
Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation.
J. Med. Genet.,
2006
Jun
, 43 (527-30).
Duchen LW
Hereditary motor end-plate disease in the mouse: light and electron microscopic studies.
J. Neurol. Neurosurg. Psychiatr.,
1970
Apr
, 33 (238-50).
Kohrman DC
et al.
Insertional mutation of the motor endplate disease (med) locus on mouse chromosome 15.
Genomics,
1995
Mar
20
, 26 (171-7).
Craner MJ
et al.
Sodium channels contribute to microglia/macrophage activation and function in EAE and MS.
Glia,
2005
Jan
15
, 49 (220-9).
Kaplan MR
et al.
Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier.
Neuron,
2001
Apr
, 30 (105-19).
Schafer DP
et al.
Early events in node of Ranvier formation during myelination and remyelination in the PNS.
Neuron Glia Biol.,
2006
May
, 2 (69-79).
Black JA
et al.
Noncanonical roles of voltage-gated sodium channels.
Neuron,
2013
Oct
16
, 80 (280-91).
Plummer NW
et al.
Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A.
Genomics,
1998
Dec
1
, 54 (287-96).
Koopmann TT
et al.
Voltage-gated sodium channels: action players with many faces.
Ann. Med.,
2006
, 38 (472-82).
Meisler MH
et al.
Sodium channelopathies in neurodevelopmental disorders.
Nat Rev Neurosci, 2021Mar, 22 (152-166).
Jones JM
et al.
Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder.
Neurobiol Dis, 2016May, 89 (36-45).
Zybura A
et al.
Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability.
Cells, 2021Jun25, 10 ().
Wittmack EK
et al.
Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase.
J. Neurosci.,
2005
Jul
13
, 25 (6621-30).
Pan Y
et al.
S-Palmitoylation of the sodium channel Nav1.6 regulates its activity and neuronal excitability.
J Biol Chem, 2020May01, 295 (6151-6164).
Fan X
et al.
Cryo-EM structure of human voltage-gated sodium channel Nav1.6.
Proc Natl Acad Sci U S A, 2023Jan31, 120 (e2220578120).
Patel RR
et al.
Human Nav1.6 Channels Generate Larger Resurgent Currents than Human Nav1.1 Channels, but the Navβ4 Peptide Does Not Protect Either Isoform from Use-Dependent Reduction.
PLoS ONE,
2015
, 10 (e0133485).
Gazina EV
et al.
Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain.
Neuroscience,
2010
Mar
10
, 166 (195-200).
Hu W
et al.
Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation.
Nat. Neurosci.,
2009
Aug
, 12 (996-1002).
Van Wart A
et al.
Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels.
J. Neurosci.,
2006
Jul
5
, 26 (7172-80).
Chen Y
et al.
Functional properties and differential neuromodulation of Na(v)1.6 channels.
Mol. Cell. Neurosci.,
2008
Aug
, 38 (607-15).
Sharkey LM
et al.
The ataxia3 mutation in the N-terminal cytoplasmic domain of sodium channel Na(v)1.6 disrupts intracellular trafficking.
J. Neurosci.,
2009
Mar
4
, 29 (2733-41).
Johannesen KM
et al.
The spectrum of intermediate SCN8A-related epilepsy.
Epilepsia, 2019May, 60 (830-844).
Talwar D
et al.
SCN8A Epilepsy, Developmental Encephalopathy, and Related Disorders.
Pediatr Neurol, 2021Sep, 122 (76-83).
Brackenbury WJ
et al.
Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth.
Proc. Natl. Acad. Sci. U.S.A.,
2010
Feb
2
, 107 (2283-8).
Barbosa C
et al.
Navβ4 regulates fast resurgent sodium currents and excitability in sensory neurons.
Mol Pain,
2015
, 11 (60).
Contributors: Katherine Johnston, Rajnish Ranjan, Michael Schartner
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/125/ , accessed on 2026 Jul 12