Nav1.5
Description: sodium channel, voltage-gated, type V, alpha subunit Gene: Scn5a Alias: nav1.5, scn5a
Nav1.5 (also known as HB1; HB2; HH1; IVF; VF1; HBBD; ICCD; LQT3; SSS1; CDCD2; CMD1E; CMPD2; PFHB1; Nav1.5), encoded by the gene scn5a, is a sodium, voltage-gated, type 5, alpha subunit channel. Nav1.5 is predominantly expressed in cardiac muscle and is responsible for the initiation of action potential and persistent and resurgent sodium current. Mutations to this channel are a cause of cardiac arrhythmias.
Experimental data
Mouse Nav1.5 gene in CHO host cells |
||
|
Click for details 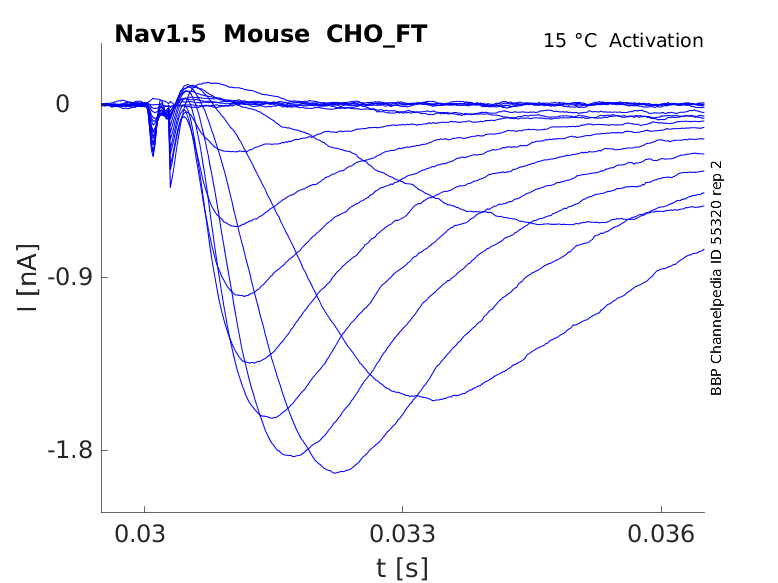
15 °Cshow 38 cells |
Click for details 
25 °Cshow 139 cells |
Click for details 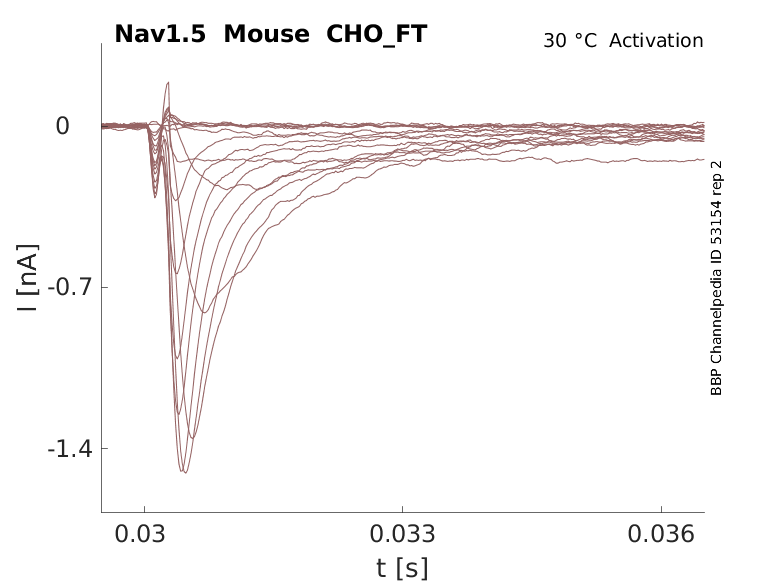
35 °Cshow 110 cells |
scn5a, the coding gene for Nav1.5, consists of 28 exons, 26 of which are coding and exon 1 being non-coding. In humans, it is located on chromosome 3 at position 22.2.
Exons 2–28 contain the protein-coding sequence. Exon 1 and part of exon 2 further encode the 5′-untranslated region (5′-UTR), whereas exon 28 also contains the 3′-UTR [2159].
scn5a is found on the same gene cluster as scn10a (Nav1.8) and scn11a (Nav1.9). All 3 genes contain an extra exon (17b) between exons 17 and 18, which corresponds to a section on the loop between domain II and domain III. scn5a is part of a voltage gated sodium channel subfamily with scn10a and scn11a, the genes encoding Nav1.8, and Nav1.9 respectively, due to their high sequence similarity and shared resistance to the pufferfish toxin, tetrodotoxin (TTX) [2160].
There exist multiple Nav1.5 transcript variants across species as a result of alternative splicing of scn5a (see Protein Isoform table).
These can be categorized into functional (ex: Nav1.5a (canonical protein), Nav1.5c, Nav1.5d, Nav1.5e and hH1c) and non‐functional (ex: Nav1.5b, Nav1.5f) splice variants [2161]. Transcripts are also specifically distributed around the organism. For example, Nav1.5a, Nav1.5e, and Nav1.5f mRNA has been found predominantly present in the rat brain [2162].
The transcript for Nav1.5e is of particular interest and is coined as the “neonatal” variant and has been found present in human, mouse and rat heart [2163]. This variant arises from the alternative splicing of exon 6 and has been shown to be developmentally regulated, with higher expression up to early postnatal life [2164].
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_198056.3 | 8519 | |
| Mouse | NM_021544.4 | 8455 | |
| Rat | NM_013125.3 | 8446 |
The canonical form of Nav1.5 is made of 2016 amino acids and has an estimated molecular weight of 227 Kda.
There exists a number of protein isoforms that arise from the translation of the aforementioned transcript variants.
These isoforms differ not only in size and weight due to alternative splicing but also in expression and location.
Certain isoforms have been shown to be majoritarily present in the brain [2162].
More research has been dedicated to Nav1.5e, the “neonatal” isoform, as it may be involved in the progression of certain diseases (ex: cancer). Nav1.5e contains a positively charged lysine residue instead of the conserved aspartate residue at the extreme extracellular end of D1:S3 [2163]. The ‘neonatal’ isoforms was shown to have altered channel gating with slower kinetics of activation and inactivation, and a lower depolarized threshold of activation in contrast to its “adult” canonical isoform counterpart [2159] [2164].
Isoforms
Post‐translational modifications play a crucial role throughout the lifecycle of Nav1.5 channels.
Glycosylation is thought to play an important role in Nav1.5 channel gating as the protein contains multiple evolutionary conserved amino acid motifs for N-glycosylation in its extracellular domains. Although the precise molecular composition of the glycans and their attachment sites have not been determined yet, their presence in cardiac Nav1.5 and impact on channel gating have been recognized. Deglycosylation experiments demonstrated that removal of glycolysation caused depolarizing shifts in both steady state activation and inactivation. [2159]
Phosphorylation is particularly important to the proper activity of Nav1.5 as multiple kinases phosphorylate and regulate channel physiology, kinetics, expression and pathology. Kinases, such as cyclic AMP‐dependent protein kinase (PKA), protein kinase C (PKC) and calcium calmodulin‐dependent kinase II (CaMKII) are abundantly expressed in areas where Nav1.5 is colocalized. Extensive research has uncovered a large number of phosphorylation sites on which these proteins act on. Removal of phosphorylation often leads to the deregulation of the channel and the emergence of channelopathies [2161].
Protein population size is determined by a balance between protein synthesis and degradation. This process can often be controlled via ubiquitination. A ubiquitin protein ligase (NEDD4‐2) binding PY‐motif has been identified at the C‐terminus of Nav1.5. NEDD4‐2 ubiquitinates Nav1.5 at this site, thus signaling for its internalization and degradation. [2161] Patch-clamp experiments were performed on HEK cells expressing wild-type and mutant forms of both Nav1.5 and Nedd4-2. Nav1.5 current density was decreased by 65% in wild-type, whereas the PY-motif mutant channels displayed no decrease in current despite the presence of Nedd-4. In contrast, catalytically inactive Nedd4-2 had no effect, indicating that ubiquitination mediates this downregulation in current and therefore channel population. However, Nedd4-2 was not found to alter Nav1.5 whole-cell or single channel biophysical properties. [2165]
Nav1.5 is subject to palmitoylation, a reversible post-translational lipid modification. Palmitoylation increases channel availability and late sodium current activity, leading to enhanced cardiac excitability and prolonged action potential duration. In contrast, blocking palmitoylation increases closed-state channel inactivation and reduces myocyte excitability. Mutations in palmitoylation sites, lead to a significant enhancement of channel closed-state inactivation and are associated with cardiac arrhythmia.[2166]
There is some evidence that Nav1.5 may be subject to other PTMs, such as arginine methylation, N-terminal acetylation and redox regulation but further research is needed to confirm their impact on the channel [2167].
Visual Representation of Nav1.5 Structure
Methodology for visual representation of structure available here

Like all voltage gated sodium channels, Nav1.5 is made up of a single protein comprised of 4 homologous domains (D1-D4). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.5 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus deactivating the channel. Nav1.5 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of rat Nav1.5 was resolved via cryo-electron microscopy, giving us a detailed insight to the channel’s architecture. Structural resolution of the ion channel highlighted certain features:
- Firstly, Nav1.5 has weaker binding affinity to NaVβ subunits compared to other Nav ion channels. This phenomenon is likely due to steric clashes when binding to the β subunits, a result from differences in amino acid sequences.
- Second, the tetrodotoxin resistant nature of Nav1.5 is due to the presence of Cys374 and Arg377. The presence of these amino acids eliminates key aromatic side chain interactions normally found in TTX-sensitive Nav channels and leads to a 500-fold decrease in TTX binding affinity. However these residues does not alter local protein conformation [2175].
It is worth noting that the structural Cryo-EM resolution was made for a specific rat Nav1.5 with truncated N and C termini in order to optimize protein expression. These structural changes may affect the protein’s properties. Further research on an untruncated protein is required to confirm the canonical structure of Nav1.5.
The approximative size/surface of Nav1.5 can be determined via the resolved or predicted structures.
Nav1.5 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Nav1.5 displays fast activation in response to depolarization and conducts a large inward Na+ current, driving the rapid upstroke of the action potential. The channel then enters a fast inactivated state with a small fraction of the remnant current [2168]. This sodium current is called persistent non-inactivating current (late INa). Late INa has minimal contribution to the action potential under physiological conditions, but plays an important role in the pathological context. Nav1.5 then returns to its resting state after repolarization.
Though Nav1.5 is mainly known as a voltage gated activated channel, it has also been shown to be mechanosensitive, with structural changes to the cell membrane influencing channel kinetics. Early research demonstrated that Nav1.5 channels expressed in HEK cells subjected to shear stress lead to increased peak current by 27% and accelerated activation [2169]. Stretch produces persistent dose-dependent shifts in the voltage dependence of activation and inactivation due to mechanical modulation of the voltage sensors, and inactivated states are stabilized by stretch. Such nuances are of physiological and pathophysiological relevance for mechanically active organs where Nav1.5 is expressed. [2170]
Single Channel Unitary Conductance
Single channel unitary conductance is determined experimentally.
For Nav1.5, there have been recordings of unitary conductance values with some degree of variance between experiments:
Model
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Origin : HEK293 cells | Recording temperature: RT
Temperature dependence: Q10 | Formalism: Markov
States: C1, C2, O1, O2, I1, I2 | Implementation: NEURON
Simulation (Nav15_a.mod):

Membrane Systems Group 2022
Origin : Mouse scn5a | Host cell: CHO
Recording temperature: 25 C | Formalism: Hodgkin-Huxley
Gates: m, h | Implementation: NEURON
Simulation (Nav15__mCHO25c.mod):

Tissue and cellular
Nav1.5 is predominantly expressed in cardiac cells and is the main sodium channel found in the heart. Within these cardiac cells, Nav1.5 is arranged in 3 different compartments: the intercalated discs, the lateral membranes, and the T-tubules [2171] [2161]
Nav1.5 was also identified present in the gut, specifically in interstitial cells of Cajal and the smooth muscle in the jejunal small intestine that participate in gastrointestinal motility [1413].
Though not originally identified in the brain, more advanced methods have found Nav1.5 present in the following brain locations [332]:
- Cerebral cortex
- Thalamus
- Hypothalamus
- Basal ganglia
- Cerebellum
- Brain Stem
- Striatum
- Olfactory system
- Limbic system: piriform cortex, septal nuclei, the diagonal band of Broca, amygdala, and habenular nuclei [844].
Developmental
Nav1.5 is not observed in adult skeletal muscles, but it is detectable in neonatal skeletal muscles suggesting a switch in transcript expression [2172].
Regulation
The regulation of expression of Nav1.5 is a complex process as, like with most proteins, proper trafficking and activity requires interactions with more than 20 proteins in distinct membrane compartments [2173]. Details for major interactions can be found in the Interaction section
Within the different structures of the brain where it can be detected, Nav1.5 is found mainly clustered around the axon. [332]
Nav1.5 being the major sodium channel in the heart, it’s main role is the conduction of the cardiac impulse [2161]
The cardiac action potential follows a specific waveform made up of 4 phases: a rapid, depolarising upstroke (0), followed by refractory period, an early hyperpolarizing downward deflection (I), a plateau that lasts ~300 ms (II) and then a repolarization phase (III). Nav1.5 activation is responsible for the fast depolarisation in the first phase (0). Any significant changes to Nav1.5 activity may significantly impact the action potential waveform and lead to subsequent pathologies [2174]
Disease
Given its crucial role in initiating fast depolarisation in cardiac action potential, mutations in scn5a that affect Nav1.5 kinetic activity often give rise to cardiac channelopathies. Mutations are distributed in every structural component, but they are especially common in conserved regions of the voltage-sensing module, selectivity filter activation gate, and fast inactivation gate [2175]
Common cardiac diseases associated with scn5a mutations are [2176]:
- Sick sinus syndrome
- Atrial Arrhythmias
- Ventricular arrhythmias
- Long QT syndrome type 3
- Brugada syndrome
- Idiopathic ventricular fibrillation
- Cardiac conduction defect
- Dilated cardiomyopathy & heart failure
- Sudden infant death syndrome
- Arrhythmogenic right ventricular cardiomyopathy
It is worth noting that in patients with identified scn5a mutations, pathologies don’t always present themselves as a distinct specific channelopathy but rather as an “overlap syndrome” presenting a mix of symptoms of scn5a related arrhythmic syndromes [2177].
Given the Nav1.5 presence and function in the gut, mutations in scn5a are associated with increases in abdominal pain syndromes. The mechanical sensitivity of Nav1.5 may also play a role in pathophysiology of irritable bowel syndrome. A recently identified Nav1.5 mutant was found to be less responsive to shear stress than wild-type Nav1.5 [2178] [2170].
scn5a mutations and changes in expression have also been implicated in the evolution and increased invasiveness of cancers such as:
Nav1.5 is one the sodium channels that is insensitive to tetrodotoxin, though it can still be blocked by TTX at high concentrations [817]
Nav1.5 is known to interact with all 4 Navβ subunits; Navβ1 and Navβ3 subunits associate non‐covalently, while β2 and β4 subunits are linked covalently by disulfide bonds. All 4 subunits are involved in the regulation of channel kinetics, gating, surface expression and voltage dependence [2161]
Nav1.5 contains a number of binding motifs for accessory proteins whose interaction regulate cellular transport, cellular localisation, gating and etc. These proteins are generally categorized as adaptor proteins, enzymes, and regulatory proteins.
Some prominent interacting proteins are:
- Ankyrin-G, a member of a family of membrane adaptor proteins. Ankyrin-G plays a crucial role in the membrane targeting and localization of Nav1.5 as proper Nav1.5 localization in cardiomyocytes is dependent on the direct interaction of ankyrin-G with the D1SII–D1SIII. Mutations in Ankyrin-G or its Nav1.5 binding site interfere with Nav1.5 surface expression in cardiomyocytes [2159].
- Calmodulin (CaM) is a small ubiquitously expressed protein which binds/senses calcium ions. CaM affects several different properties of Nav1.5, including channel inactivation and persistent current [2181] However, contrary to CaM’s interaction with Nav1.4, CaM interaction with Nav1.5 does not lead to calcium dependent inactivation (CDI). The difference in calcium sensing properties is likely due to the post-IQ region (Δpost-IQ), where CaM binds to Nav1.5, as removal of the wild type Δpost-IQ does lead to Nav1.5 CDI [2181] [2182].
For additional resources on potential drug and compound interactions:
- Known and predicted drug interactions with Nav1.5
- Known and predicted animal toxin interactions with Nav1.5
References
Onkal R
et al.
Alternative splicing of Nav1.5: an electrophysiological comparison of 'neonatal' and 'adult' isoforms and critical involvement of a lysine residue.
J. Cell. Physiol.,
2008
Sep
, 216 (716-26).
Wu L
et al.
Localization of Nav1.5 sodium channel protein in the mouse brain.
Neuroreport,
2002
Dec
20
, 13 (2547-51).
Catterall WA
et al.
International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels.
Pharmacol. Rev.,
2005
Dec
, 57 (397-409).
Fozzard HA
et al.
Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms.
Physiol. Rev.,
1996
Jul
, 76 (887-926).
Gao R
et al.
Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer.
Oncol. Rep.,
2010
May
, 23 (1293-9).
Brisson L
et al.
Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae.
Oncogene,
2011
Apr
28
, 30 (2070-6).
Hartmann HA
et al.
Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain.
Nat. Neurosci.,
1999
Jul
, 2 (593-5).
Lyford GL
et al.
Ion channels in gastrointestinal smooth muscle and interstitial cells of Cajal.
,
2003
Dec
, 3 (583-7).
Zhang J
et al.
N-type fast inactivation of a eukaryotic voltage-gated sodium channel.
Nat Commun, 20220517, 13 (2713).
Rook MB
et al.
Biology of cardiac sodium channel Nav1.5 expression.
Cardiovasc. Res.,
2012
Jan
1
, 93 (12-23).
Mancino AS
et al.
Spliced isoforms of the cardiac Nav1.5 channel modify channel activation by distinct structural mechanisms.
J Gen Physiol, 2022May02, 154 ().
Iqbal SM
et al.
Phosphorylation of cardiac voltage-gated sodium channel: Potential players with multiple dimensions.
Acta Physiol (Oxf), 2019Mar, 225 (e13210).
Wang J
et al.
Multiple Nav1.5 isoforms are functionally expressed in the brain and present distinct expression patterns compared with cardiac Nav1.5.
Mol Med Rep, 2017Jul, 16 (719-729).
Chioni AM
et al.
A novel polyclonal antibody specific for the Na(v)1.5 voltage-gated Na(+) channel 'neonatal' splice form.
J. Neurosci. Methods,
2005
Sep
30
, 147 (88-98).
Murphy LL
et al.
Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia.
Heart Rhythm,
2012
Apr
, 9 (590-7).
Van Bemmelen MX
et al.
Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination.
Circ. Res.,
2004
Aug
6
, 95 (284-91).
Pei Z
et al.
Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation.
Nat Commun, 2016Jun23, 7 (12035).
Marionneau C
et al.
Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications.
J. Mol. Cell. Cardiol.,
2015
May
, 82 (36-47).
Zhang Z
et al.
Kinetic model of Nav1.5 channel provides a subtle insight into slow inactivation associated excitability in cardiac cells.
PLoS ONE,
2013
, 8 (e64286).
Strege PR
et al.
Cytoskeletal modulation of sodium current in human jejunal circular smooth muscle cells.
Am. J. Physiol., Cell Physiol.,
2003
Jan
, 284 (C60-6).
Beyder A
et al.
Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel.
J. Physiol. (Lond.),
2010
Dec
15
, 588 (4969-85).
Gellens ME
et al.
Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel.
Proc. Natl. Acad. Sci. U.S.A.,
1992
Jan
15
, 89 (554-8).
Kallen RG
et al.
Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle.
Neuron,
1990
Feb
, 4 (233-42).
Shy D
et al.
Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model.
Biochim. Biophys. Acta,
2013
Apr
, 1833 (886-94).
Takla M
et al.
The cardiac CaMKII-Nav1.5 relationship: From physiology to pathology.
J Mol Cell Cardiol, 2020Feb, 139 (190-200).
Porretta AP
et al.
SCN5A overlap syndromes: An open-minded approach.
Heart Rhythm, 2022Aug, 19 (1363-1368).
Saito YA
et al.
Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy.
Am. J. Physiol. Gastrointest. Liver Physiol.,
2009
Feb
, 296 (G211-8).
Gradek F
et al.
Sodium Channel Nav1.5 Controls Epithelial-to-Mesenchymal Transition and Invasiveness in Breast Cancer Cells Through its Regulation by the Salt-Inducible Kinase-1.
Sci Rep, 2019Dec09, 9 (18652).
Guzel RM
et al.
Colorectal cancer invasiveness in vitro: Predominant contribution of neonatal Nav1.5 under normoxia and hypoxia.
J Cell Physiol, 2019May, 234 (6582-6593).
Wang C
et al.
Structural analyses of Ca²⁺/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation.
Nat Commun,
2014
, 5 (4896).
Nathan S
et al.
Structural basis of cytoplasmic NaV1.5 and NaV1.4 regulation.
J Gen Physiol, 2021Jan04, 153 ().
Contributors: Katherine Johnston
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/124/ , accessed on 2026 Aug 02