Nav1.1
Description: sodium channel, voltage-gated, type I, alpha Gene: Scn1a Alias: nav1.1, scn1a1
Nav1.1, encoded by the gene scn1a, is a sodium, voltage-gated, type I, alpha subunit channel. Nav1.1 is widely expressed in the CNS and to a lesser extent in the PNS. It is involved in the generation of the action potential. Mutations in the channel are often the cause of epileptic disorders.
scn1a is the coding gene for Nav1.1. In humans, scn1a is located on chromosome 2q24 and is made up of 28 exons, 26 of which are coding and exons 1 and 2 being non-coding. scn1a spans a total length of 6030 nucleotides. scn1a is present in the same cluster as the genes coding for the other voltage gated sodium channels scn2a (nav1.2) and scn3a (nav1.3) [2120]
There exist multiple Nav1.1 transcript variants across species as a result of alternative splicing of scn1a (see Protein Isoform table).
The best studied scn1a splicing variants are often referred to as the adult and neonatal forms, although both forms are expressed in adults. They result from the expression of either exon 5A (adult) or 5N (neonatal). Both transcripts of exon 5 encode nearly identical sequences in domain I [2233].
scn1a is also alternatively spliced in exon 11, which codes for a region within the
first intracellular loop.
Exon 20 of scn1a contains multiple splice sites and that produce a number of variants that lead to either truncated isoforms in Domain III or isoforms with a structurally distinct pore loop in the same region. This splicing event is similar to the one that occurs for exon 18 in scn8a (Nav1.6). [2234] [2235]
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_001165963.4 | 11940 | |
| Mouse | NM_001313997.1 | 8320 | |
| Rat | NM_030875.2 | 8397 |
The human Nav1.1 protein is composed of 2009 amino acid (aa) and has a molecular weight of 229 Kda.
There exists a number of protein isoforms that arise from the translation of the aforementioned transcript variants:
- Nav1.1A and Nav1.1N isoforms are nearly identical in sequences. However, there are three amino acid changes in the extracellular loop between S3 and S4 of domain I that differ between the two exons (adult to neonatal): Y203F, D207N, and V211F. [2234]
- The alternative splicing event at exon 11 generates two shortened isoforms, one which lacks 11 amino acids and the other 28 amino acids respectively. The functional consequence of exon 11 splicing is unknown. [2234]
- Exon 20 (“poison exon”, PE) contains in-frame stop codons that result in a truncated channel protein. This exon is generally incorporated in the proteins transcribed in non-neuronal cells, thus preventing the expression of the ion channel [2186]. In neuronal cells, PEs are preferentially skipped to make a full-length transcript and fully functioning canonical isoform [2236]
Isoforms
Nav1.1 contains a number of post translational modification (PTM) sites, indicating that it is likely subject to PTMs, such as phosphorylation and glycosylation. Nav1.1 was shown to also be subjected to arginine methylation. However, the exact timing of PTMs and their impact on channel kinetics or expression have not been extensively studied. [2233]
Visual Representation of Nav1.1 Structure
Methodology for visual representation of structure available here
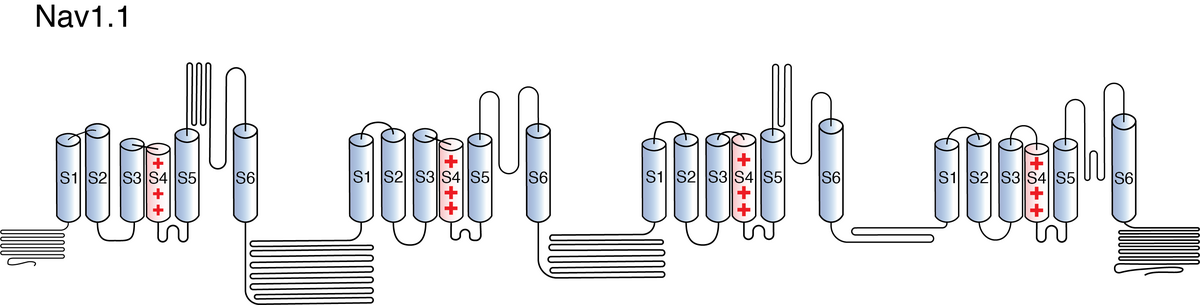
Like all voltage gated sodium channels, Nav1.1 is made up of a single protein comprised of 4 homologous domains (DI-DIV). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.1 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus inactivating the channel. Nav1.1 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of human Nav1.1, in complex with Navβ4, was resolved via cryo-electron microscopy, giving us a detailed insight to the specific structural features of Nav1.1. The study did highlight areas within the channel considered as mutational hotspots for many channelopathies. The majority of the mutations were concentrated on the extracellular loops above the pore domain and the supporting segments of the selectivity filter, as well as the pore domain itself and the voltage-sensing domains. The two former impair the structural integrity of the channel whereas the latter 2 interfere with electromechanical couple and fast inactivation [2237]
Nav1.1 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Nav1.1, like most voltage gated sodium channels, is characterized by its fast kinetics.
The channel has a low threshold of activation, fast activation and inactivation, and slow recovery from inactivation.
Nav1.1 therefore activates rapidly in response to depolarization and conducts a large inward Na+ current, driving the rapid upstroke of the action potential. Fast inactivation quickly kicks in following the channel opening, with the protein returning to its resting state after repolarization. [2238]
Single channel unitary conductance
Single channel unitary conductance is determined experimentally.
For Nav1.1, there has been a recording of unitary conductance value:
- 17 pS [2239]
Model
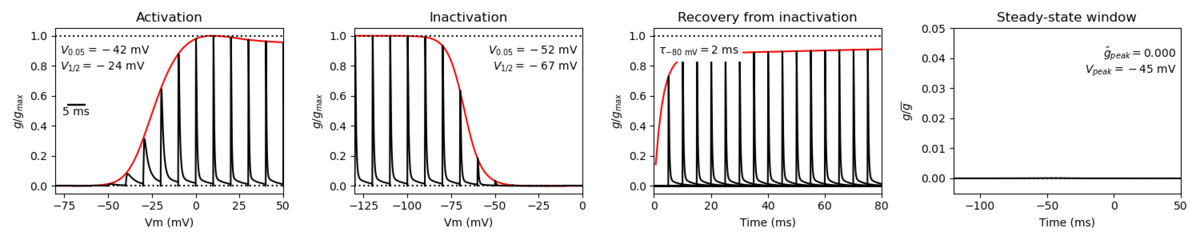
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Species : Human | Gene: scn1a (+ β1, β2)
Host cell: tsa201 (HEK293) | Temperature: RT (to 25 C by Q10)
Formalism: Markov | States: C1, C2, O1, O2, I1, I2
Implementation: NEURON | Simulation: Nav11_a.mod
Membrane Systems Group 2023
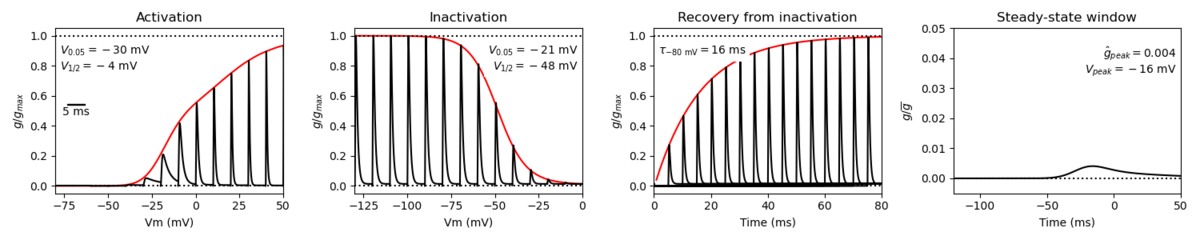
Species: Mouse | Gene: Scn1a
Host cell: CHO | Temperature: 25 C
Formalism: Hodgkin-Huxley | Gates: m3, h
Implementation: NEURON | Simulation: Nav11__mCHO25c.mod
Tissue and Cellular
Nav1.1 is predominantly expressed in the central nervous system (CNS), in regions such as the hippocampus and the cerebral cortex [2240]
Nav1.1 is also expressed in the peripheral nervous system (PNS), where it is primarily found in myelinated medium-diameter sensory neurons. [2241]
Nav1.1 can also be found in non-excitable cells, such as keratinocytes and microglia [2117]
Developmental
Nav1.1 is expressed at low-levels at birth and increases postnatally, continuing so into adulthood [2101]
Nav1.1 is predominantly found in the soma of most neuronal cells, more specifically at the axon initial segment (AIS) in both the CNS and PNS. The channel has also been localized at the nodes of Ranvier in certain areas of the brain and the spinal cord. High channel densities for either location are dependent on cell population [2118]
Action Potential Generation
Given its main location at the neuron soma, Nav1.1 is thought to control neuron excitability via the integration of synaptic impulses and then setting the action potential initiation threshold. In its other locations, it is responsible for the propagation of the action potential [820]
Nociceptions
Nav1.1 is thought to play a role in nociception. Some studies have demonstrated that activation or sensitisation of Nav1.1 expressing fiber led to increased pain in mice models. Blockade of Nav1.1 reversed this phenomenon, eliminating pain [2241] [2241]
Channelopathies
Epilepsies
Given its important presence in the brain and its role in AP generation, one of the most infamous consequences of Nav1.1 deregulation is the development of epilepsy disorders. Indeed, functional mutations of the channel lead to gain-of-function or loss-of-function mutations, causing a number of channelopathies:
- Epilepsy [1373]
- Febrile epilepsy [1373]
- Generalized epilepsy with febrile seizure (GEFS+) [1373]
- Dravet syndrome [1373]
- Doose syndrome [1374]
- Rasmussen's encephalitis [1375]
- Lennox-Gastaut syndrome [1376]
- Panayiotopoulos syndrome [1379]
Autism
Targeted DNA sequencing from autism spectrum disorders in the brain identified de novo mutations in scn1a, suggesting that deregulation of the channel may play a role in the development of such disorders [2242] [2243].
Migraines
Mutations in scn1a are known to cause familial hemiplegic migraine (FHM), an autosomal dominant inherited subtype of severe migraines [39].
Alzheimer's disease (AD)
Nav1.1 may have an indirect impact on the development of Alzheimer’s disease. Early evidence shows that overexpression of Nav1.1 in certain interneurons led to improvements in cognitive function in human amyloid precursor protein (hAPP)-transgenic mice, which simulate key aspects of AD. Conversely, Nav1.1 deficient interneurons lead to behavioral abnormalities in wild type mice [2244] [1381]. Though multiple different factors may contribute to the development of AD, Nav1.1 could be a potential target for future treatment development.
Nav1.1 is known to interact with many different compounds and accessory proteins that modify its properties and activity.[2245]
Nav1.1 is TTX-sensitive. Its activity is completely blocked by application of nanomolar concentrations of tetrodotoxin [1376]
Calmodulin and Ca++ Concentration
Nav1.1 can be regulated by CaM and by Ca++ alone, both compounds interacting directly with the C-terminal region of the channel.
CaM was shown to accelerate the inactivation kinetics of Nav1.1 and induce a left shift in voltage-dependent activation.
Increasing Ca++ concentrations lead to a right shift in the voltage-dependence of fast inactivation and a slowing down of the fast inactivation kinetics. For high Ca++ concentrations, this effect competed with the acceleration induced by CaM alone.
Interestingly, the depolarizing action of calcium antagonized the hyperpolarizing shift of the voltage-dependence of activation due to CaM overexpression [482].
Beta subunits [2246]
Nav1.1 is reported to interact and be regulated by all four Navβ subunits, Navβ1 to Navβ4, with each subunit having a different effect on the channel’s properties:
- Navβ1 accelerates fast inactivation
- Navβ2 accelerates fast inactivation & increases persistent current
- Navβ3 increases persistent current
- Navβ4 increases activation and non-activating current.
FGF
Fibroblast Growth Factor Homologous Factors (FHF1-4) modulate sodium channels in an isoform-dependent manner. In fact, FHF4 (B isoform) reduces current density by 90% of Nav1.1. [1372]
- Known and predicted drug interactions with Nav1.1
- Known and predicted animal toxin interactions with Nav1.1
References
Kalume F
et al.
Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy.
J. Neurosci.,
2007
Oct
10
, 27 (11065-74).
Spampanato J
et al.
Increased neuronal firing in computer simulations of sodium channel mutations that cause generalized epilepsy with febrile seizures plus.
J. Neurophysiol.,
2004
May
, 91 (2040-50).
Barela AJ
et al.
An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability.
J. Neurosci.,
2006
Mar
8
, 26 (2714-23).
Cestèle S
et al.
Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel.
J. Neurosci.,
2008
Jul
16
, 28 (7273-83).
Rusconi R
et al.
Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant.
J. Neurosci.,
2007
Oct
10
, 27 (11037-46).
Ogiwara I
et al.
Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation.
J. Neurosci.,
2007
May
30
, 27 (5903-14).
Duflocq A
et al.
Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments.
Mol. Cell. Neurosci.,
2008
Oct
, 39 (180-92).
Garrido JJ
et al.
A targeting motif involved in sodium channel clustering at the axonal initial segment.
Science,
2003
Jun
27
, 300 (2091-4).
Leipold E
et al.
Combinatorial interaction of scorpion toxins Lqh-2, Lqh-3, and LqhalphaIT with sodium channel receptor sites-3.
Mol. Pharmacol.,
2004
Mar
, 65 (685-91).
Gaudioso C
et al.
Calmodulin and calcium differentially regulate the neuronal Nav1.1 voltage-dependent sodium channel.
,
2011
Jun
25
, ().
Trimmer JS
et al.
Localization of voltage-gated ion channels in mammalian brain.
Annu. Rev. Physiol.,
2004
, 66 (477-519).
Han S
et al.
Na(V)1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms.
Proc. Natl. Acad. Sci. U.S.A.,
2012
Feb
7
, 109 (E368-77).
Westenbroek RE
et al.
Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons.
Neuron,
1989
Dec
, 3 (695-704).
Whitaker WR
et al.
Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum.
J. Comp. Neurol.,
2000
Jun
19
, 422 (123-39).
Lou JY
et al.
Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels.
J. Physiol. (Lond.),
2005
Nov
15
, 569 (179-93).
Escayg A
et al.
Sodium channel SCN1A and epilepsy: mutations and mechanisms.
Epilepsia,
2010
Sep
, 51 (1650-8).
Yordanova I
et al.
One novel Dravet syndrome causing mutation and one recurrent MAE causing mutation in SCN1A gene.
Neurosci. Lett.,
2011
Apr
25
, 494 (180-3).
Ohmori I
et al.
Rasmussen encephalitis associated with SCN 1 A mutation.
Epilepsia,
2008
Mar
, 49 (521-6).
Savio-Galimberti E
et al.
Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies.
Front Pharmacol,
2012
, 3 (124).
Kahlig KM
et al.
Divergent sodium channel defects in familial hemiplegic migraine.
Proc. Natl. Acad. Sci. U.S.A.,
2008
Jul
15
, 105 (9799-804).
Weiss LA
et al.
Sodium channels SCN1A, SCN2A and SCN3A in familial autism.
Mol. Psychiatry,
2003
Feb
, 8 (186-94).
Grosso S
et al.
SCN1A mutation associated with atypical Panayiotopoulos syndrome.
Neurology,
2007
Aug
7
, 69 (609-11).
Verret L
et al.
Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model.
Cell,
2012
Apr
27
, 149 (708-21).
Han S
et al.
Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission.
Nature,
2012
Sep
20
, 489 (385-90).
Yu FH
et al.
Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy.
Nat. Neurosci.,
2006
Sep
, 9 (1142-9).
Cheah CS
et al.
Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome.
Proc. Natl. Acad. Sci. U.S.A.,
2012
Sep
4
, 109 (14646-51).
Cantrell AR
et al.
Neuromodulation of Na+ channels: an unexpected form of cellular plasticity.
Nat. Rev. Neurosci.,
2001
Jun
, 2 (397-407).
Smith RS
et al.
Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development.
Neuron,
2018
Sep
05
, 99 (905-913.e7).
Black JA
et al.
Noncanonical roles of voltage-gated sodium channels.
Neuron,
2013
Oct
16
, 80 (280-91).
Wang J
et al.
Distribution and function of voltage-gated sodium channels in the nervous system.
Channels (Austin), 2017Nov02, 11 (534-554).
Ademuwagun IA
et al.
Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions.
Front Neurol, 2021, 12 (600050).
Meisler MH
et al.
Sodium channelopathies in neurodevelopmental disorders.
Nat Rev Neurosci, 2021Mar, 22 (152-166).
Onwuli DO
et al.
An update on transcriptional and post-translational regulation of brain voltage-gated sodium channels.
Amino Acids, 2016Mar, 48 (641-651).
Fletcher EV
et al.
Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker.
J. Biol. Chem.,
2011
Oct
21
, 286 (36700-8).
Oh Y
et al.
Novel splice variants of the voltage-sensitive sodium channel alpha subunit.
Neuroreport,
1998
May
11
, 9 (1267-72).
Carvill GL
et al.
Poison exons in neurodevelopment and disease.
Curr Opin Genet Dev, 2020Dec, 65 (98-102).
Pan X
et al.
Comparative structural analysis of human Nav1.1 and Nav1.5 reveals mutational hotspots for sodium channelopathies.
Proc Natl Acad Sci U S A, 2021Mar16, 118 ().
Benarroch EE
Signaling molecules of the CNS as targets of autoimmunity.
Handb Clin Neurol,
2016
, 133 (17-38).
Cooper EC
(What to do) when epilepsy gene mutations stop making sense.
Epilepsy Curr, 2007Jan-Feb, 7 (23-5).
Osteen JD
et al.
Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain.
Nature, 2016Jun23, 534 (494-9).
D'Gama AM
et al.
Targeted DNA Sequencing from Autism Spectrum Disorder Brains Implicates Multiple Genetic Mechanisms.
Neuron,
2015
Dec
2
, 88 (910-7).
O'Roak BJ
et al.
Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations.
Nature,
2012
May
10
, 485 (246-50).
Martinez-Losa M
et al.
Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer's Disease.
Neuron, 2018Apr04, 98 (75-89.e5).
Vormstein-Schneider D
et al.
Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans.
Nat Neurosci, 2020Dec, 23 (1629-1636).
Aman TK
et al.
Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels.
J. Neurosci.,
2009
Feb
18
, 29 (2027-42).
Contributors: Katherine Johnston, Rajnish Ranjan, Michael Schartner
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/120/ , accessed on 2026 Jun 29