Nav1.2
Description: sodium channel, voltage-gated, type II, alpha 1 Gene: Scn2a Alias: nav1.2, scn2a1
Nav1.2, encoded by the gene scn2a, is a sodium, voltage-gated, type 2, alpha subunit channel. Nav1.2 is predominantly expressed in the CNS. It is involved in the control of backpropagation and peaking of the action potential. Mutations in the channel are often the cause of epileptic disorders.
The human gene for Nav1.2, scn2a, is made up of 27 exons, 26 of which are coding and exons 1 being non-coding. scn2a is located on chromosome 2 (2q24.3). It is found on the same gene cluster as other voltage gated sodium channels: Nav1.1 (scn1a) and Nav1.3 (scn3a). This common gene location is consistent with evidence of a common evolutionary origin [817]
There exist multiple Nav1.2 transcript variants across species as a result of the alternative splicing of scn2a (see Protein Isoform table).
The best studied scn2a splicing variants are referred to as the adult and neonatal forms. They result from the alternative mRNA splicing of either exon 5A (adult) or 5N (neonatal).
Both variants have almost identical coding regions, with only a 36 nucleotide difference. The expression of either variant seems to be mutually exclusive and the selection of these two exons are developmentally regulated (see Expression & Distribution) [2247] [2248]
| Species | NCBI accession | Length (nt) | |
|---|---|---|---|
| Human | NM_021007.3 | 8630 | |
| Mouse | NM_001099298.3 | 8819 | |
| Rat | NM_012647.2 | 8556 |
The human Nav1.2 protein is composed of 2005 amino acid (aa) and has a molecular weight of 228 Kda.
The Nav1.2 isoforms that result from the alternative splicing of exon 5A and 5N differ by one amino acid at position 209: an asparagine in Nav1.2 (5N) and an aspartic acid in Nav1.2 (5A). This difference is located in the S3-S4 helix in domain I of the channel. [2272]
Functionally, the adult isoform of the channel is more active than the neonatal one. Nav1.2 (5A) showed a hyperpolarized shift of the voltage dependence of activation, a depolarized shift of the voltage dependence of fast inactivation, a slower time course, and an accelerated recovery from fast inactivation in comparison to the neonatal splice variant. However, the functional importance of this difference remains unclear. It has been suggested that the neonatal isoform might reduce the neuronal excitability and therefore have a protective role against seizures during early development [2249] [2248]
Isoforms
Nav1.2 contains a number of glycosylation sites, indicating that the protein is likely glycosylated. However, the exact timing of glycosylation and its impacts on channel kinetics or expression have not been extensively studied (uniprot)
Nav1.2 is subject to phosphorylation by various enzymes [2145]:
- PKC was show to both inhibit peak current and slow macroscopic fast inactivation.
- CaMKII phosphorylation of Nav1.2 increases persistent sodium currents and excitability.
- Fast inactivation of Nav1.2 channels is regulated via tyrosine phosphorylation by Fyn kinase and dephosphorylation by receptor phosphoprotein tyrosine phosphatase-beta [2250] [2251]
Nav1.2 is subject to ubiquitination as it possesses a PY motif and was shown to be negatively regulated by Nedd4-2. [2122]
Sumoylation is the addition of a small ubiquitin-like modifier (SUMO) protein by an enzyme. Nav1.2 is likely to be regulated by sumoylation on dedicated binding sites, as addition of SUMO protein leads to an increase in Nav1.2 mediated current expressed in HEK cells. [2252]
Palmitoylation sites were identified on Nav1.2. Lipid modification on these sites has been shown to regulate and modify channel gating and pharmacological properties. [2145] [2253]
There is some evidence that Nav1.2 may be subject to other PTMs, such as redox regulation. For example, Chloramine-T can impair the inactivation of Nav1.2 [2144]
Visual Representation of Nav1.2 Structure
Methodology for visual representation of structure available here
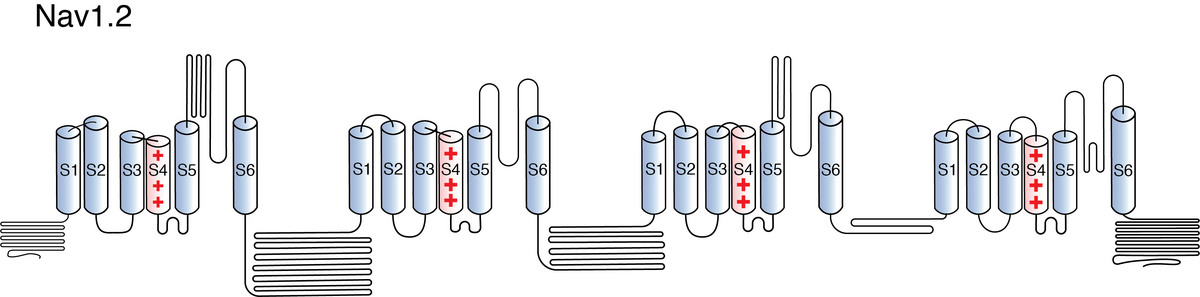
Like all voltage gated sodium channels, Nav1.2 is made up of a single protein comprised of 4 homologous domains (D1-D4). Each domain is made up of 6 transmembrane subunits (S1-S6). S1-4 form the voltage sensing domain (VSD) whereas the S5-6 form the pore module (PM). The S4 subunit of each domain contains a series of positively charged residues. When membrane depolarization occurs, these charged residues cause the movement of the S4 subunit, inducing a conformational change in S5-S6, opening of the channel and allowing the entry of sodium ions into the cell. Soon after opening, rapid inactivation of Nav1.2 is instigated by the binding of the IFM motif, found in the loop between D3 and D4, to a hydrophobic receptor site next to the S6 in D4. This binding causes the shift of S6, allosterically closing the channel, thus deactivating the channel. Nav1.2 then returns to its resting state following the hyperpolarization of the cell membrane [2115].
The structure of rat Nav1.2 was resolved via cryo-electron microscopy to an overall resolution of 3.0 angstroms, giving us a detailed insight to the channel’s architecture. Structural resolution of the ion channel highlighted certain features such as how certain Navβ subunits interact with the pore domain through disulphide bonds and which specific Nav1.2 residues, in the extracellular segment of the channel’s selectivity filter, interact with KIIIA, a Nav blocker. [2254]
Nav1.2 predicted AlphaFold size
Methodology for AlphaFold size prediction and disclaimer are available here
Nav1.2 displays similar kinetics to other voltage-gated sodium channels, namely fast activation and fast inactivation kinetics. However, the channel does have its own distinct current properties. Nav1.2 displays more depolarized activation and thus requires substantial depolarization for activation. However, Nav1.2 shows a greater accumulation of inactivation at higher frequencies of stimulation (20-100 Hz) and thus are likely to generate lower frequencies of firing. [51] [2255]
It is worth noting that kinetics of the channel may change depending on the influence of the expressed isoform (see Isoforms)
Single channel unitary conductance
Single channel unitary conductance is determined experimentally.
For Nav1.2, single channel unitary conductance has yet to be determined.
Model
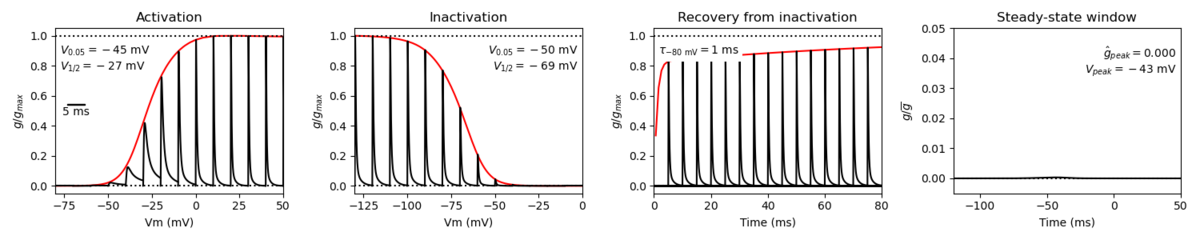
A single kinetic model for all human voltage-gated sodium channels (Balbi et al, 2017)
https://modeldb.science/230137
Species : Human | Gene: scn2a (+ β1,β2)
Host cell: tsa201 (HEK293) | Temperature: RT (to 25 C by Q10)
Formalism: Markov | States: C1, C2, O1, O2, I1, I2
Implementation: NEURON | Simulation: Nav12_a.mod
Membrane Systems Group 2023
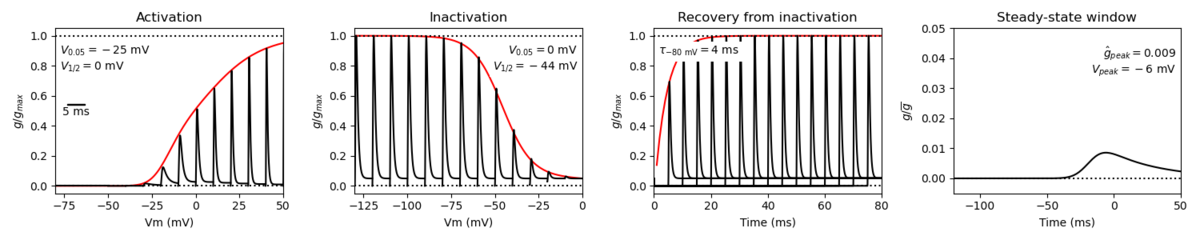
Species: Mouse | Gene: Scn2a
Host cell: CHO | Temperature: 25 C
Formalism: Hodgkin-Huxley | Gates: m3, h
Implementation: NEURON | Simulation: Nav12__mCHO25c.mod
Tissue and Cellular
Nav1.2 is predominantly expressed in the CNS and has been detected at high levels in the cerebral cortex, the cerebellum, the hippocampus, and the caudate. source. In the cerebral cortex, NaV1.2 is found in glutamatergic pyramidal cells rather than in inhibitory neurons. [2256]
Nav1.2 can also be found in non-neuronal cells, such as astrocytes, fibroblasts, islet β-cells, odontoblasts, osteoblasts, and schwann cells where they also play important roles in cellular function. [2257]
Developmental
Nav1.2 is the main voltage gated sodium channel expressed during early developpement. In the first postnatal week in mice, corresponding to late gestation through to the first year of life in humans, Nav1.2 is the only sodium channel isoform expressed in the axon and axon initiation segment (AIS) and is thus solely responsible for the initiation and propagation of APs. Later in development, NaV1.2 in the axon and distal AIS is replaced by NaV1.6. [2258]
This switch in developmental expression coincides with the differential expression of Nav1.2 neonatal and adult isoforms. Nav1.2(5N) mRNAs are relatively abundant at birth but are gradually replaced by Nav1.2 (5A) mRNAs as development progresses. In the mouse cortex, neonatal splice mRNA is present in approximately two-fold excess compared with the canonical adult expressed Nav1.2(5A) transcript during early development, but drops to one third of the canonical splice variant within the first 2 weeks of life. [2272]
However, there is some variation in developmental expression between isoforms, species, and brain regions. For example, in rats, Nav1.2 mRNA levels increase steadily until the first postnatal week and then maintain high levels in adulthood in rostral regions. In caudal regions and the spinal cord, mRNA levels steadily increase during development before decreasing to low levels after the second postnatal week. Conversely, the cerebellum shows low levels of Nav1.2 during the first 2 postnatal weeks but high expression levels in adults [2259]
During early development, Nav1.2 is present all across the axon initial segment (AIS) and the nodes of Ranvier before being replaced by Nav1.6.
Later in development and adulthood, Nav1.2 is predominantly found at the proximal AIS, the closest point to the soma. [365] [2255]
Somatodendritic excitability
As it is the main voltage gated sodium channel present, Nav1.2 is the main driver of action potential initiation during early development. The channel is then replaced by Nav1.6 over the course of later development and this developmentally regulated expression may be one mechanism that counters the normally high excitability of neonatal neurons and helps to reduce seizure susceptibility in normal human infants.[417]
In adult cells, Nav1.2’s main roles are to control action potential backpropagation, by setting a threshold for action potential backpropagation to the soma and dendrites, and to help determine the height of the AP peak.
Control of Action Potential Peak
Nav1.2’s activation threshold is higher than that of other voltage gated sodium channels found around the AIS, namely Nav1.6. Nav1.2 is also found at the proximal AIS, the site nearest to the soma. Upon depolarisation, Nav1.6 “threshold” channels open first, at lower potentials, causing the start of the AP. As Nav1.2 activates at higher potential and thus later during AP generation, this stops the AP from immediately propagating back “up” the neuron. However, once activated, the channel contributes to somatodendritic excitability, thus setting the threshold for the generation of somatodendritic potentials. Removal of Nav1.2 leads to a failure in backpropagation and impaired dendritic excitability. [2260][2258] [2194]
Nav1.2 channels are also particularly permeable to Ca2+ and mediate Ca2+ influx in the AIS during the AP [2261]. This permeability allows for the activation of BK CAKCs in the AIS. BK channels regulate the peak and the early phase of the AP repolarization, shaping the AP waveform at the site of generation. Some evidence shows that a decrease in Nav1.2 current leads to a decrease in BK current, resulting in a widening of the early phase of AP repolarization. Therefore the interplay between Nav1.2 and BK channels may be a major determinant of the AP peak and a key regulator of the AP shape. [2260]
Channelopathies
Given Nav1.2’s important roles within neuronal cells, particularly during development, mutations to the protein often result in biophysical alterations of the channel. Gain of function mutations generally lead to hyperpolarized shifts in activation and faster channel activation, depolarised shifts in inactivation, accelerated recovery from fast inactivation, increase persistent current, and increased current amplitude. Loss of function mutations, conversely, result in to depolarized shifts in activation and slower channel activation, hyperpolarized shifts in inactivation, slowed recovery from fast inactivation, and decreased current amplitude [2248]
These mutations manifest themselves as a number of disorders, including:
- Famillial autism [1378]
- Autism spectrum disorders [1387]
- Neonatal epilepsy [1388]
- Late ataxia [1388]
- Myoclonus [1388]
- Pain [1388]
- Perinatal lethal [1389]
- Multiple sclerosis [404]
- GEFS+ [2262]
- BFNIS [2263]
- Intellectual disability [2258]
There is also early evidence that deregulation of Nav1.2 in the hippocampus causes intellectual disability, namely impaired spatial memory, and scn2a has been frequently identified in brain metastases [2264] [2265]
TTX
Nav1.2 is TTX-sensitive. Its activity is completely blocked by application of nanomolar concentrations of tetrodotoxin [1376]. Nav1.2 also interacts with a number of compounds and accessory protein that modify its properties and alter its activity.
Ankyrin G
Ankyrin G interacts with Nav1.2 and anchors the protein to the plasma membrane.[412]
Beta Subunit
Relative to NaV1.2 alone, Nav1.2 + Navβ1 currents have right-shifted voltage dependence of activation, fast and slow inactivation and reduced use dependence. In addition, the NaV1.2 + Navβ1 current entered fast inactivation slightly faster than Nav1.2 channels alone.[416]
Navβ2 only causes a small left shift in voltage dependance of activation and its binding is likely to play a role in other aspects of protein function (expression, trafficking, etc.)
Navβ3, like Navβ1, causes a positive shift in the voltage dependence of activation
Coexpression of Navβ4 with Nav1.2 results in a negative shift in the voltage dependence of channel activation. [406]
REST
Repressor element-1 silencing transcription/neuron-restrictive silencer factor (REST/NRSF) is a transcriptional regulator that represses the expression of Nav1.2, particularly in non-neuronal cells [2266] [2267]
Contactin
Contactin, a glycosyl-phosphatidylinositol (GPI)-anchored predominantly neuronal cell surface glycoprotein, associates with Nav1.2 and enhances the density of the channel on the plasma membrane [2268]
When expressed alongside contactin, Nav1.2 cells was shown to have threefold to fourfold higher peak sodium currents than cells with Nav1.2 alone [2269]
Calmodulin & Ca2+
Calmodulin is a small ubiquitous eukaryotic Ca2+-sensing regulatory protein. CaM binds to Nav1.2 via the IQ motif in the C-terminal end of the protein and is thought to regulate the channel’s activity in response to changes in calcium concentration. However further research is needed to determine the exact impact of the interaction on Nav1.2 [2270]
- Known and predicted drug interactions with Nav1.2
- Known and predicted animal toxin interactions with Nav1.2
References
Shah BS
et al.
Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta3, in rat CNS.
J. Physiol. (Lond.),
2001
Aug
1
, 534 (763-76).
Rush AM
et al.
Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones.
J. Physiol. (Lond.),
2005
May
1
, 564 (803-15).
Tan M
et al.
Effects of BmK AS on Nav1.2 expressed in Xenopus laevis oocytes.
Cell Biol. Toxicol.,
2008
Apr
, 24 (143-9).
Misra SN
et al.
Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures.
Epilepsia,
2008
Sep
, 49 (1535-45).
Osorio N
et al.
Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells.
J. Physiol. (Lond.),
2010
Feb
15
, 588 (651-70).
Craner MJ
et al.
Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger.
Proc. Natl. Acad. Sci. U.S.A.,
2004
May
25
, 101 (8168-73).
Rasband MN
et al.
Dysregulation of axonal sodium channel isoforms after adult-onset chronic demyelination.
J. Neurosci. Res.,
2003
Aug
15
, 73 (465-70).
Yu FH
et al.
Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2.
J. Neurosci.,
2003
Aug
20
, 23 (7577-85).
Qin N
et al.
Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit.
Eur. J. Biochem.,
2003
Dec
, 270 (4762-70).
Garrido JJ
et al.
A targeting motif involved in sodium channel clustering at the axonal initial segment.
Science,
2003
Jun
27
, 300 (2091-4).
Rasband MN
et al.
Paranodal transverse bands are required for maintenance but not initiation of Nav1.6 sodium channel clustering in CNS optic nerve axons.
Glia,
2003
Nov
, 44 (173-82).
Chabbert C
et al.
Voltage-gated Na+ channel activation induces both action potentials in utricular hair cells and brain-derived neurotrophic factor release in the rat utricle during a restricted period of development.
J. Physiol. (Lond.),
2003
Nov
15
, 553 (113-23).
Garrido JJ
et al.
Dynamic compartmentalization of the voltage-gated sodium channels in axons.
Biol. Cell,
2003
Oct
, 95 (437-45).
McEwen DP
et al.
Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin.
J. Biol. Chem.,
2004
Apr
16
, 279 (16044-9).
Osorio N
et al.
Differential targeting and functional specialization of sodium channels in cultured cerebellar granule cells.
J. Physiol. (Lond.),
2005
Dec
15
, 569 (801-16).
Hossain WA
et al.
Where is the spike generator of the cochlear nerve? Voltage-gated sodium channels in the mouse cochlea.
J. Neurosci.,
2005
Jul
20
, 25 (6857-68).
de Ruiter MM
et al.
Voltage-gated sodium channels in cerebellar Purkinje cells of mormyrid fish.
J. Neurophysiol.,
2006
Jul
, 96 (378-90).
Xu R
et al.
Generalized epilepsy with febrile seizures plus-associated sodium channel beta1 subunit mutations severely reduce beta subunit-mediated modulation of sodium channel function.
Neuroscience,
2007
Aug
10
, 148 (164-74).
Xu R
et al.
A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel.
Mol. Cell. Neurosci.,
2007
Jun
, 35 (292-301).
Huth T
et al.
Four-mode gating model of fast inactivation of sodium channel Nav1.2a.
Pflugers Arch.,
2008
Oct
, 457 (103-19).
Zhu MM
et al.
The alpha-like scorpion toxin BmK I enhances membrane excitability via persistent sodium current by preventing slow inactivation and deactivation of rNav1.2a expressed in Xenopus Oocytes.
Toxicol In Vitro,
2009
Jun
, 23 (561-8).
Berendt FJ
et al.
Multi-site Phosphorylation of Voltage-Gated Sodium Channel alpha Subunits from Rat Brain.
,
2010
Feb
5
, ().
Gordon D
et al.
Tissue-specific expression of the RI and RII sodium channel subtypes.
Proc. Natl. Acad. Sci. U.S.A.,
1987
Dec
, 84 (8682-6).
Catterall WA
et al.
International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels.
Pharmacol. Rev.,
2005
Dec
, 57 (397-409).
Westenbroek RE
et al.
Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons.
Neuron,
1989
Dec
, 3 (695-704).
Weiss LA
et al.
Sodium channels SCN1A, SCN2A and SCN3A in familial autism.
Mol. Psychiatry,
2003
Feb
, 8 (186-94).
Cantrell AR
et al.
Neuromodulation of Na+ channels: an unexpected form of cellular plasticity.
Nat. Rev. Neurosci.,
2001
Jun
, 2 (397-407).
Sanders SJ
et al.
De novo mutations revealed by whole-exome sequencing are strongly associated with autism.
Nature,
2012
May
10
, 485 (237-41).
Liao Y
et al.
SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain.
Neurology,
2010
Oct
19
, 75 (1454-8).
Planells-Cases R
et al.
Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice.
Biophys. J.,
2000
Jun
, 78 (2878-91).
Eijkelkamp N
et al.
Neurological perspectives on voltage-gated sodium channels.
Brain,
2012
Sep
, 135 (2585-612).
Boiko T
et al.
Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon.
Neuron,
2001
Apr
, 30 (91-104).
Rougier JS
et al.
Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins.
Am. J. Physiol., Cell Physiol.,
2005
Mar
, 288 (C692-701).
Kassmann M
et al.
Oxidation of multiple methionine residues impairs rapid sodium channel inactivation.
Pflugers Arch.,
2008
Sep
, 456 (1085-95).
Pei Z
et al.
Posttranslational Modification of Sodium Channels.
Handb Exp Pharmacol, 2018, 246 (101-124).
Hu W
et al.
Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation.
Nat. Neurosci.,
2009
Aug
, 12 (996-1002).
Sarao R
et al.
Developmentally regulated alternative RNA splicing of rat brain sodium channel mRNAs.
Nucleic Acids Res.,
1991
Oct
25
, 19 (5673-9).
Hedrich UBS
et al.
SCN2A channelopathies: Mechanisms and models.
Epilepsia, 2019Dec, 60 Suppl 3 (S68-S76).
Muller GK
The neonatal SCN2A mutant channel mimics adult channel properties.
J Gen Physiol, 2020May04, 152 ().
Beacham D
et al.
Sites and molecular mechanisms of modulation of Na(v)1.2 channels by Fyn tyrosine kinase.
J. Neurosci.,
2007
Oct
24
, 27 (11543-51).
Ahn M
et al.
Regulation of Na(v)1.2 channels by brain-derived neurotrophic factor, TrkB, and associated Fyn kinase.
J. Neurosci.,
2007
Oct
24
, 27 (11533-42).
Plant LD
et al.
SUMOylation of NaV1.2 channels mediates the early response to acute hypoxia in central neurons.
Elife, 2016Dec28, 5 ().
Bosmans F
et al.
Palmitoylation influences the function and pharmacology of sodium channels.
Proc. Natl. Acad. Sci. U.S.A.,
2011
Dec
13
, 108 (20213-8).
Pan X
et al.
Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA.
Science, 2019Mar22, 363 (1309-1313).
Kaczmarek LK
Loss of NaV1.2-Dependent Backpropagating Action Potentials in Dendrites Contributes to Autism and Intellectual Disability.
Neuron, 2019Aug21, 103 (551-553).
Berret E
et al.
Oligodendroglial excitability mediated by glutamatergic inputs and Nav1.2 activation.
Nat Commun, 2017Sep15, 8 (557).
Spratt PWE
et al.
The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex.
Neuron, 2019Aug21, 103 (673-685.e5).
Beckh S
et al.
Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development.
EMBO J.,
1989
Dec
1
, 8 (3611-6).
Filipis L
et al.
Nav1.2 and BK channel interaction shapes the action potential in the axon initial segment.
J Physiol, 2023May, 601 (1957-1979).
Hanemaaijer NA
et al.
Ca2+ entry through NaV channels generates submillisecond axonal Ca2+ signaling.
Elife, 2020Jun17, 9 ().
Sugawara T
et al.
A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction.
Proc. Natl. Acad. Sci. U.S.A.,
2001
May
22
, 98 (6384-9).
Scalmani P
et al.
Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures.
J. Neurosci.,
2006
Oct
4
, 26 (10100-9).
Middleton SJ
et al.
Altered hippocampal replay is associated with memory impairment in mice heterozygous for the Scn2a gene.
Nat Neurosci, 2018Jul, 21 (996-1003).
Sun J
et al.
Genomic signatures reveal DNA damage response deficiency in colorectal cancer brain metastases.
Nat Commun, 2019Jul18, 10 (3190).
Armisén R
et al.
Repressor element-1 silencing transcription/neuron-restrictive silencer factor is required for neural sodium channel expression during development of Xenopus.
J. Neurosci.,
2002
Oct
1
, 22 (8347-51).
Ballas N
et al.
Regulation of neuronal traits by a novel transcriptional complex.
Neuron,
2001
Aug
16
, 31 (353-65).
Rush AM
et al.
Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive sodium channels in DRG neurons.
Eur. J. Neurosci.,
2005
Jul
, 22 (39-49).
Kazarinova-Noyes K
et al.
Contactin associates with Na+ channels and increases their functional expression.
J. Neurosci.,
2001
Oct
1
, 21 (7517-25).
Feldkamp MD
et al.
Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage-dependent sodium channel.
Structure,
2011
May
11
, 19 (733-47).
Thompson CH
et al.
Alternative splicing potentiates dysfunction of early-onset epileptic encephalopathy SCN2A variants.
J Gen Physiol, 2020Mar02, 152 ().
Contributors: Katherine Johnston, Rajnish Ranjan, Michael Schartner
To cite this page: [Contributors] Channelpedia https://channelpedia.epfl.ch/wikipages/121/ , accessed on 2026 Jul 17